Bioinformatics and Machine Learning (BML) Lab, Department of Electrical Engineering and Computer Science (EECS), University of Missouri-Columbia, Columbia, MO, USA.
Stowers Institute for Medical Research, Kansas City, MO, USA.
Sci Rep. 2021 Jun 10;11(1):12295. doi: 10.1038/s41598-021-91827-7.
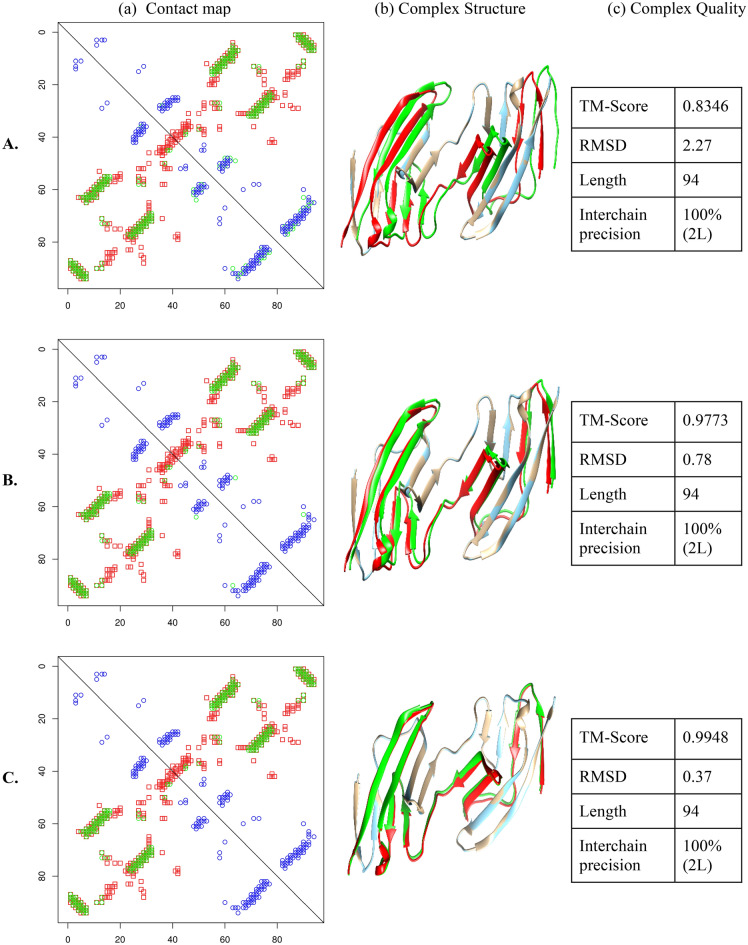
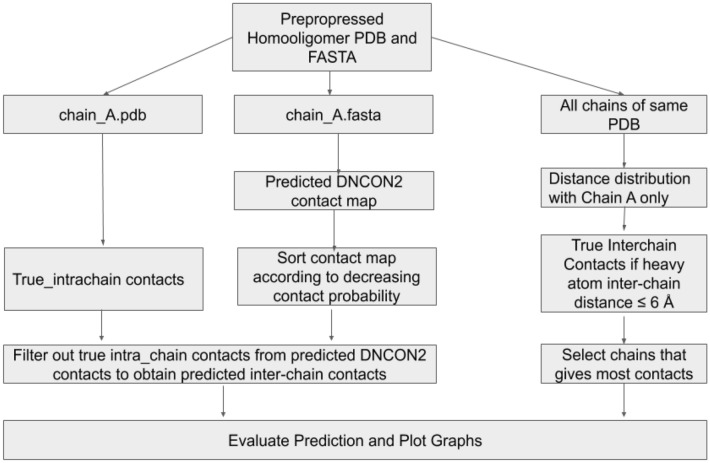
Deep learning methods that achieved great success in predicting intrachain residue-residue contacts have been applied to predict interchain contacts between proteins. However, these methods require multiple sequence alignments (MSAs) of a pair of interacting proteins (dimers) as input, which are often difficult to obtain because there are not many known protein complexes available to generate MSAs of sufficient depth for a pair of proteins. In recognizing that multiple sequence alignments of a monomer that forms homomultimers contain the co-evolutionary signals of both intrachain and interchain residue pairs in contact, we applied DNCON2 (a deep learning-based protein intrachain residue-residue contact predictor) to predict both intrachain and interchain contacts for homomultimers using multiple sequence alignment (MSA) and other co-evolutionary features of a single monomer followed by discrimination of interchain and intrachain contacts according to the tertiary structure of the monomer. We name this tool DNCON2_Inter. Allowing true-positive predictions within two residue shifts, the best average precision was obtained for the Top-L/10 predictions of 22.9% for homodimers and 17.0% for higher-order homomultimers. In some instances, especially where interchain contact densities are high, DNCON2_Inter predicted interchain contacts with 100% precision. We also developed Con_Complex, a complex structure reconstruction tool that uses predicted contacts to produce the structure of the complex. Using Con_Complex, we show that the predicted contacts can be used to accurately construct the structure of some complexes. Our experiment demonstrates that monomeric multiple sequence alignments can be used with deep learning to predict interchain contacts of homomeric proteins.
深度学习方法在预测链内残基残基接触方面取得了巨大成功,已被应用于预测蛋白质之间的链间接触。然而,这些方法需要输入一对相互作用的蛋白质(二聚体)的多重序列比对(MSA),这通常很难获得,因为没有很多已知的蛋白质复合物可用于生成足够深度的 MSA 一对蛋白质。在认识到形成同源多聚体的单体的多重序列比对包含了链内和链间残基对接触的共进化信号后,我们应用了 DNCON2(一种基于深度学习的蛋白质链内残基残基接触预测器),使用单体的多重序列比对(MSA)和其他共进化特征来预测同源多聚体的链内和链间接触,然后根据单体的三级结构来区分链间和链内接触。我们将这个工具命名为 DNCON2_Inter。允许在两个残基位移内进行真正的阳性预测,对于同源二聚体的 Top-L/10 预测,最佳平均精度为 22.9%,对于更高阶的同源多聚体,最佳平均精度为 17.0%。在某些情况下,特别是当链间接触密度较高时,DNCON2_Inter 可以预测链间接触的准确率达到 100%。我们还开发了 Con_Complex,这是一种复杂结构重建工具,它使用预测的接触来产生复合物的结构。使用 Con_Complex,我们表明可以使用预测的接触来准确构建一些复合物的结构。我们的实验表明,单体的多重序列比对可以与深度学习一起用于预测同源蛋白质的链间接触。