MacDonell Ryan J, Dickerson Claire E, Birch Clare J T, Kumar Alok, Edmunds Claire L, Biercuk Michael J, Hempel Cornelius, Kassal Ivan
School of Chemistry, University of Sydney NSW 2006 Australia
University of Sydney Nano Institute, University of Sydney NSW 2006 Australia.
Chem Sci. 2021 Jun 18;12(28):9794-9805. doi: 10.1039/d1sc02142g. eCollection 2021 Jul 21.
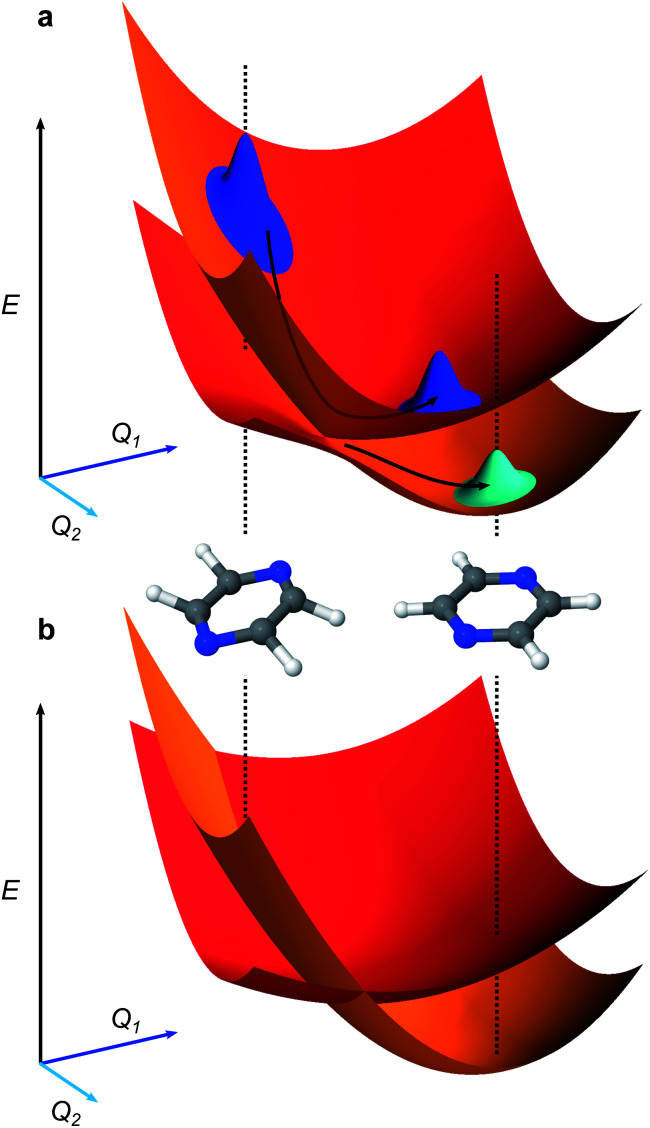
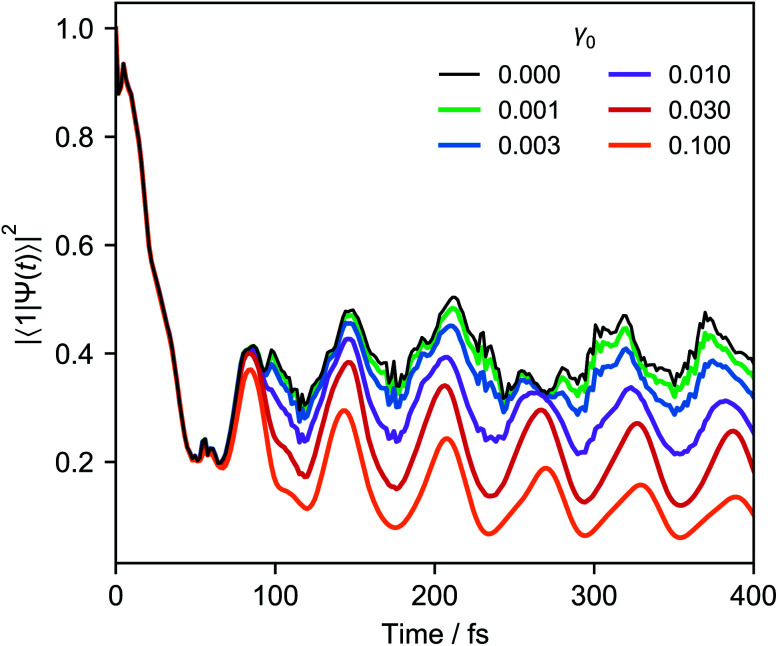
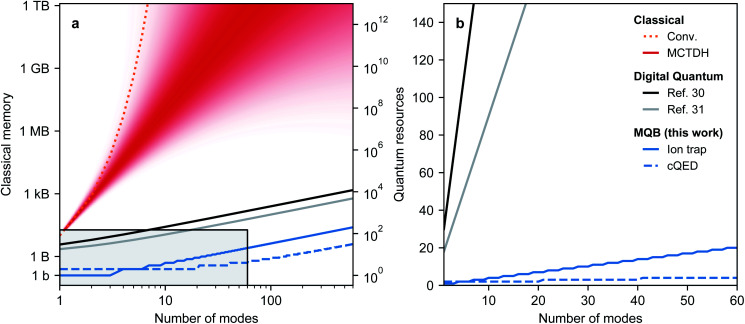
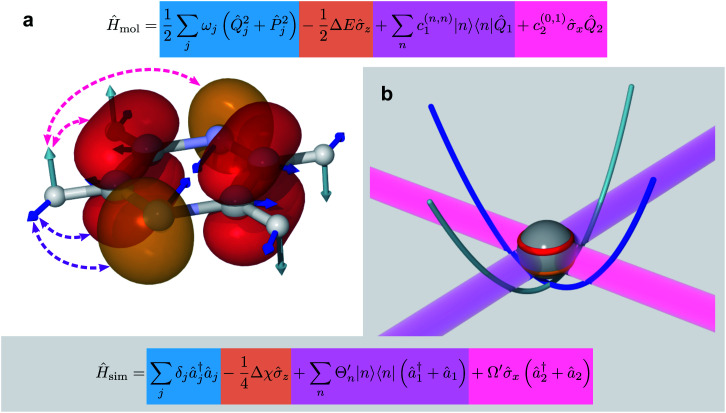
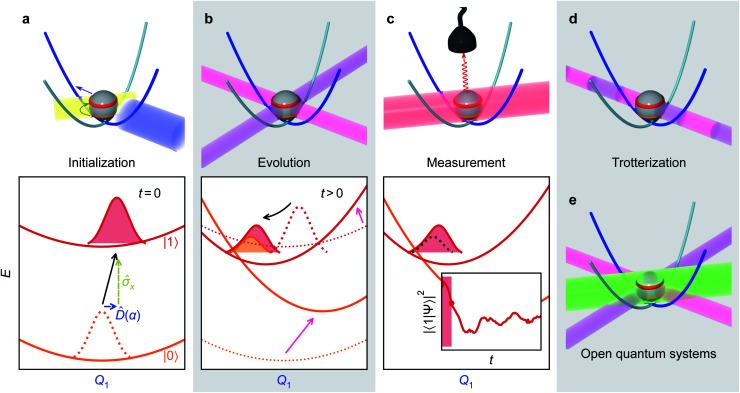
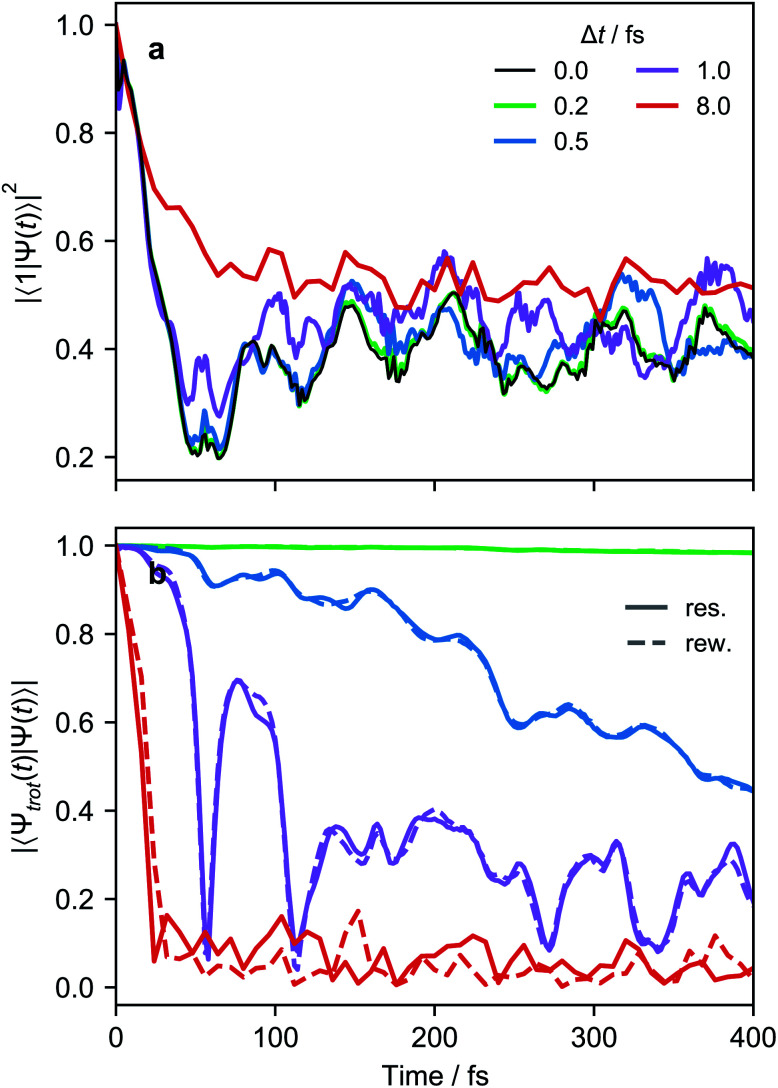
Ultrafast chemical reactions are difficult to simulate because they involve entangled, many-body wavefunctions whose computational complexity grows rapidly with molecular size. In photochemistry, the breakdown of the Born-Oppenheimer approximation further complicates the problem by entangling nuclear and electronic degrees of freedom. Here, we show that analog quantum simulators can efficiently simulate molecular dynamics using commonly available bosonic modes to represent molecular vibrations. Our approach can be implemented in any device with a qudit controllably coupled to bosonic oscillators and with quantum hardware resources that scale linearly with molecular size, and offers significant resource savings compared to digital quantum simulation algorithms. Advantages of our approach include a time resolution orders of magnitude better than ultrafast spectroscopy, the ability to simulate large molecules with limited hardware using a Suzuki-Trotter expansion, and the ability to implement realistic system-bath interactions with only one additional interaction per mode. Our approach can be implemented with current technology; , the conical intersection in pyrazine can be simulated using a single trapped ion. Therefore, we expect our method will enable classically intractable chemical dynamics simulations in the near term.
超快化学反应难以模拟,因为它们涉及纠缠的多体波函数,其计算复杂度会随着分子大小迅速增加。在光化学中,玻恩-奥本海默近似的失效通过纠缠核自由度和电子自由度,进一步使问题复杂化。在此,我们表明模拟量子模拟器可以使用常见的玻色子模式来表示分子振动,从而有效地模拟分子动力学。我们的方法可以在任何具有可控耦合到玻色子振荡器的量子位以及量子硬件资源与分子大小成线性缩放的设备中实现,并且与数字量子模拟算法相比,可显著节省资源。我们方法的优点包括时间分辨率比超快光谱法好几个数量级,能够使用铃木- Trotter展开在有限硬件条件下模拟大分子,以及能够仅通过每个模式增加一种相互作用来实现实际的系统-浴相互作用。我们的方法可以用当前技术实现;例如,吡嗪中的锥形交叉点可以用单个俘获离子来模拟。因此,我们预计我们的方法将在短期内实现经典方法难以处理的化学动力学模拟。