Velasco-Vélez Juan-Jesús, Carbonio Emilia A, Chuang Cheng-Hao, Hsu Cheng-Jhih, Lee Jyh-Fu, Arrigo Rosa, Hävecker Michael, Wang Ruizhi, Plodinec Milivoj, Wang Feng Ryan, Centeno Alba, Zurutuza Amaia, Falling Lorenz J, Mom Rik Valentijn, Hofmann Stephan, Schlögl Robert, Knop-Gericke Axel, Jones Travis E
Department of Heterogeneous Reactions, Max Planck Institute for Chemical Energy Conversion, Mülheim an der Ruhr 45470, Germany.
Department of Inorganic Chemistry, Fritz-Haber-Institut der Max-Planck-Gesellschaft, Berlin 14195, Germany.
J Am Chem Soc. 2021 Aug 18;143(32):12524-12534. doi: 10.1021/jacs.1c01655. Epub 2021 Aug 6.
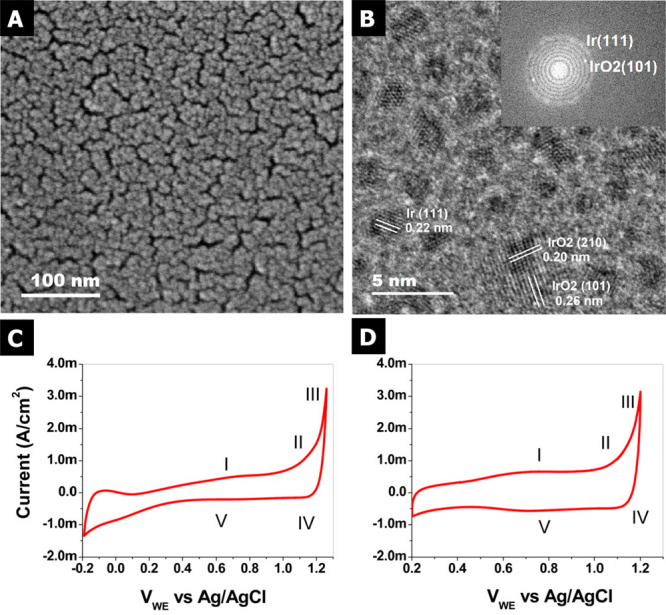
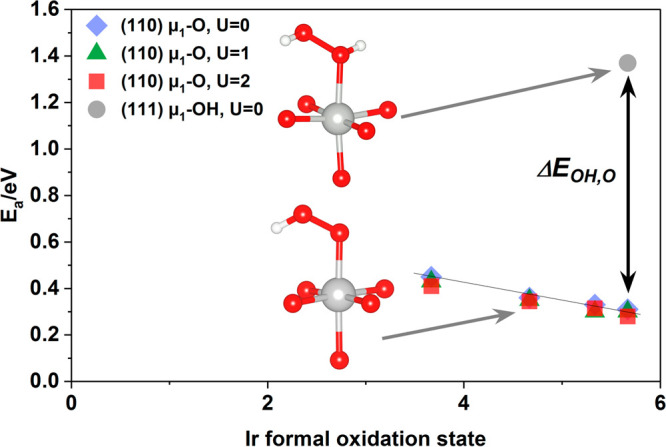
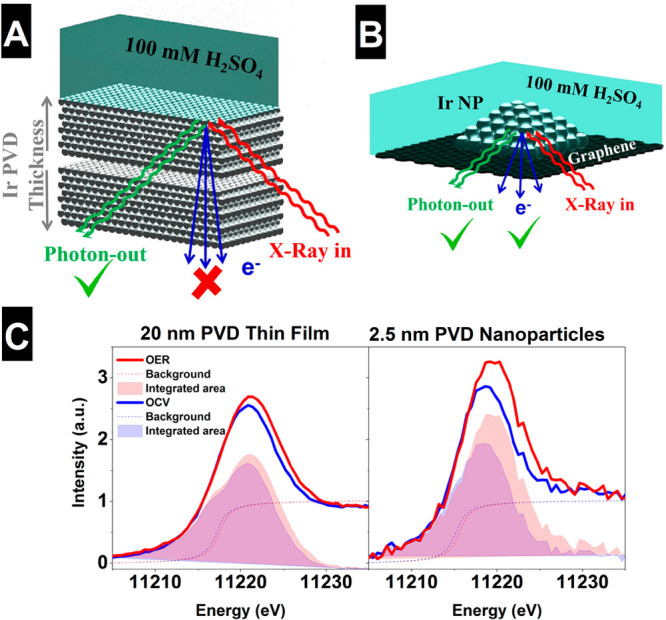
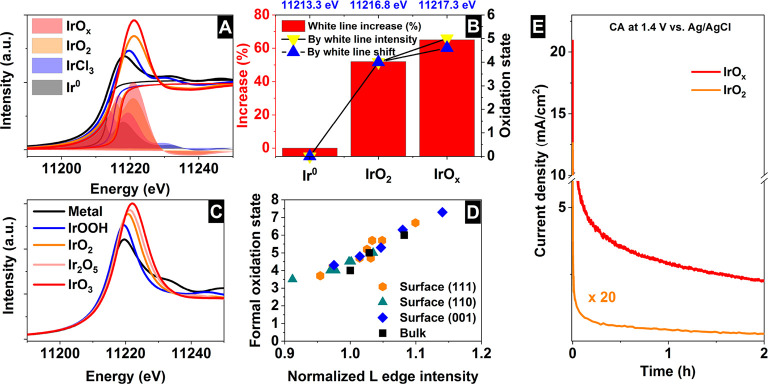
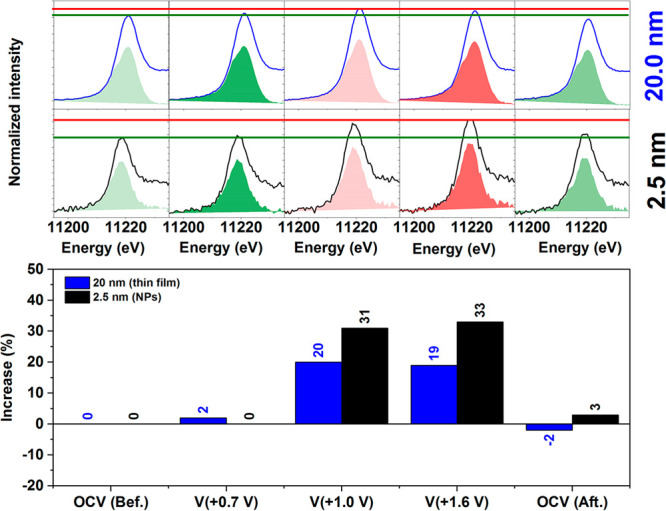
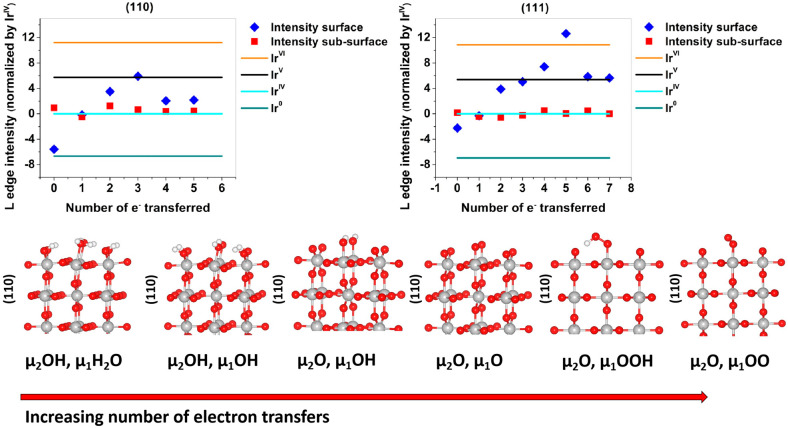
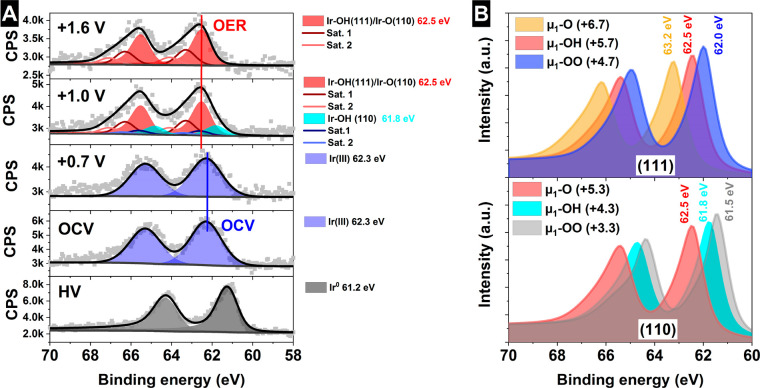
Iridium and ruthenium and their oxides/hydroxides are the best candidates for the oxygen evolution reaction under harsh acidic conditions owing to the low overpotentials observed for Ru- and Ir-based anodes and the high corrosion resistance of Ir-oxides. Herein, by means of cutting edge surface and bulk sensitive X-ray spectroscopy techniques, specifically designed electrode nanofabrication and DFT calculations, we were able to reveal the electronic structure of the active IrO centers (i.e., oxidation state) during electrocatalytic oxidation of water in the surface and bulk of high-performance Ir-based catalysts. We found the oxygen evolution reaction is controlled by the formation of empty Ir 5d states in the surface ascribed to the formation of formally Ir species leading to the appearance of electron-deficient oxygen species bound to single iridium atoms (μ-O and μ-OH) that are responsible for water activation and oxidation. Oxygen bound to three iridium centers (μ-O) remains the dominant species in the bulk but do not participate directly in the electrocatalytic reaction, suggesting bulk oxidation is limited. In addition a high coverage of a μ-OO (peroxo) species during the OER is excluded. Moreover, we provide the first photoelectron spectroscopic evidence in bulk electrolyte that the higher surface-to-bulk ratio in thinner electrodes enhances the material usage involving the precipitation of a significant part of the electrode surface and near-surface active species.
铱和钌及其氧化物/氢氧化物是在苛刻酸性条件下析氧反应的最佳候选材料,这是由于基于钌和铱的阳极具有较低的过电位以及氧化铱具有高耐腐蚀性。在此,借助前沿的表面和体相灵敏X射线光谱技术、专门设计的电极纳米制造技术以及密度泛函理论计算,我们得以揭示高性能铱基催化剂的表面和体相中水电催化氧化过程中活性IrO中心的电子结构(即氧化态)。我们发现析氧反应受表面空的Ir 5d态形成的控制,这归因于形式上的Ir物种的形成,导致与单个铱原子结合的缺电子氧物种(μ-O和μ-OH)出现,这些物种负责水的活化和氧化。与三个铱中心结合的氧(μ-O)在体相中仍然是主要物种,但不直接参与电催化反应,这表明体相氧化受到限制。此外,在析氧反应期间排除了μ-OO(过氧)物种的高覆盖率。此外,我们在本体电解质中提供了首个光电子能谱证据,即较薄电极中较高的表面与体相之比提高了材料利用率,这涉及电极表面和近表面活性物种的大量沉淀。