John A. Paulson School of Engineering and Applied Sciences, Harvard University, Cambridge, MA, USA.
Broad Institute of MIT and Harvard, Cambridge, MA, USA.
Nat Commun. 2021 Oct 8;12(1):5909. doi: 10.1038/s41467-021-26044-x.
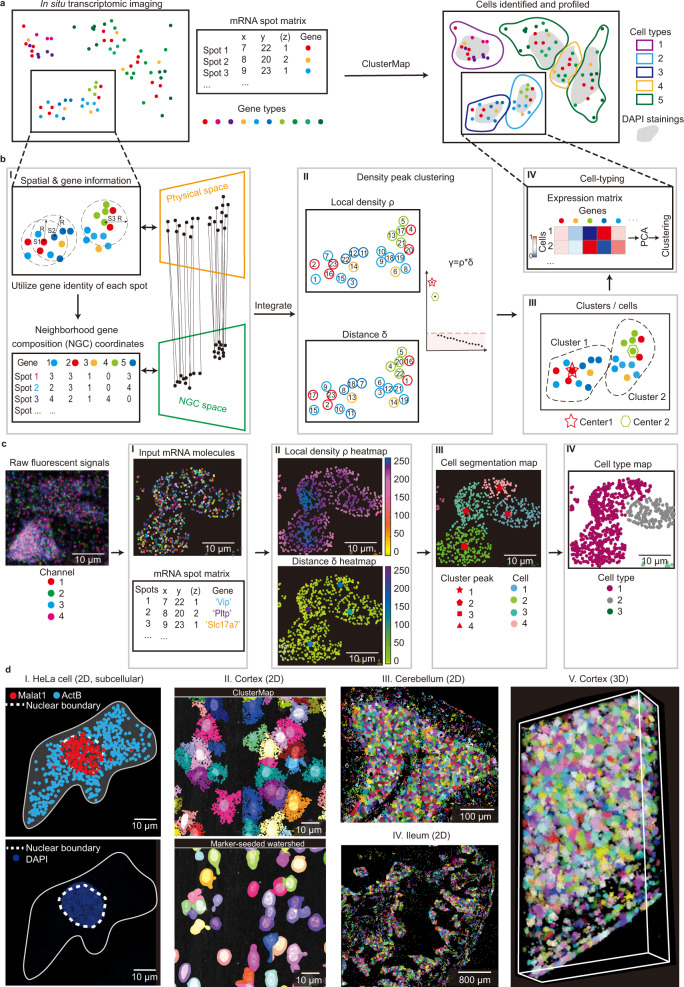
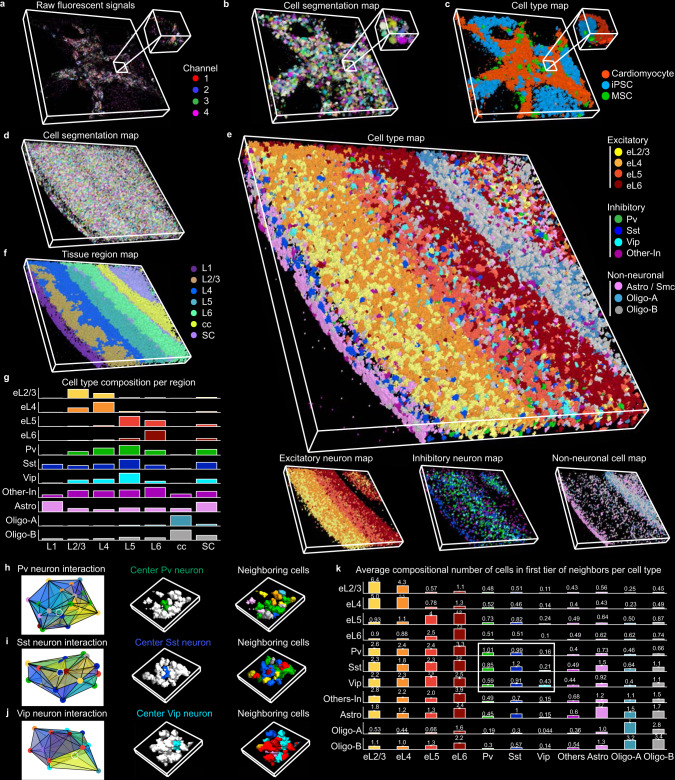
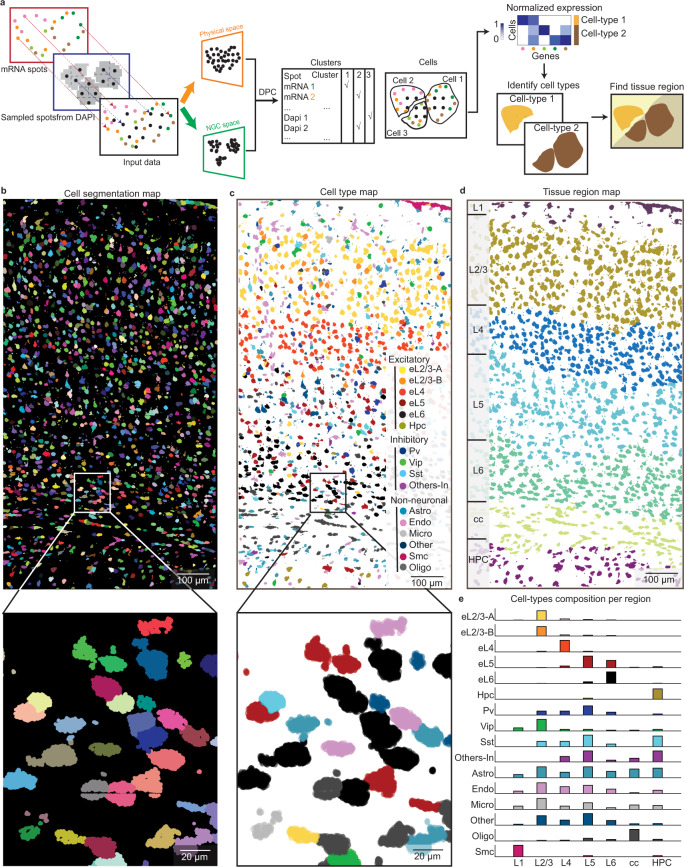
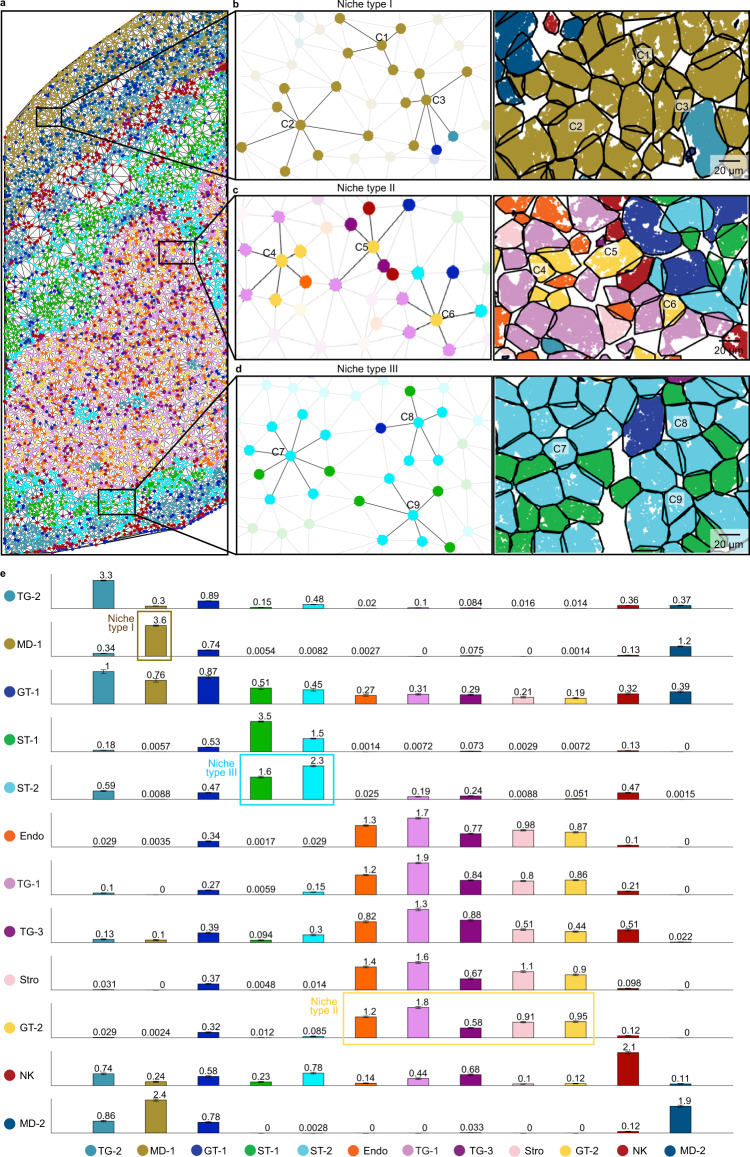
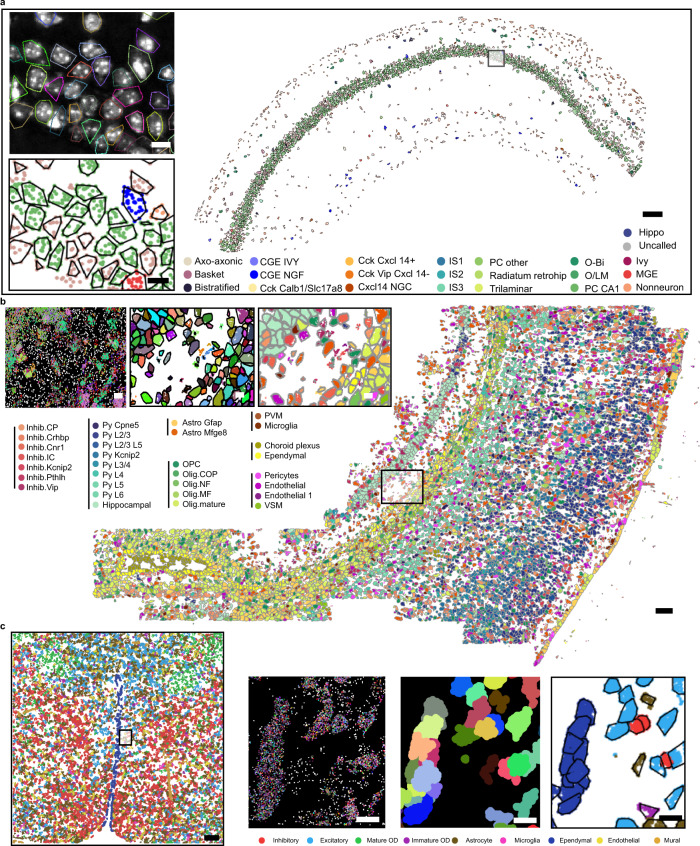
Quantifying RNAs in their spatial context is crucial to understanding gene expression and regulation in complex tissues. In situ transcriptomic methods generate spatially resolved RNA profiles in intact tissues. However, there is a lack of a unified computational framework for integrative analysis of in situ transcriptomic data. Here, we introduce an unsupervised and annotation-free framework, termed ClusterMap, which incorporates the physical location and gene identity of RNAs, formulates the task as a point pattern analysis problem, and identifies biologically meaningful structures by density peak clustering (DPC). Specifically, ClusterMap precisely clusters RNAs into subcellular structures, cell bodies, and tissue regions in both two- and three-dimensional space, and performs consistently on diverse tissue types, including mouse brain, placenta, gut, and human cardiac organoids. We demonstrate ClusterMap to be broadly applicable to various in situ transcriptomic measurements to uncover gene expression patterns, cell niche, and tissue organization principles from images with high-dimensional transcriptomic profiles.
定量分析空间背景中的 RNA 对于理解复杂组织中的基因表达和调控至关重要。原位转录组学方法可在完整组织中生成空间分辨的 RNA 图谱。然而,目前缺乏用于综合分析原位转录组学数据的统一计算框架。在这里,我们提出了一种无监督且无需注释的框架,称为 ClusterMap,它结合了 RNA 的物理位置和基因身份,将任务表述为点模式分析问题,并通过密度峰聚类(DPC)识别具有生物学意义的结构。具体来说,ClusterMap 可以精确地将 RNA 聚类到亚细胞结构、细胞体和组织区域中,无论是在二维还是三维空间中,并且在包括小鼠大脑、胎盘、肠道和人类心脏类器官在内的多种组织类型上表现一致。我们证明了 ClusterMap 可以广泛应用于各种原位转录组学测量,从具有高维转录组谱的图像中揭示基因表达模式、细胞生态位和组织组织原则。