Program in Computational & Molecular Biophysics, Washington University School of Medicine, Saint Louis, Missouri 63110, United States.
Center for Computing Research, Sandia National Laboratories, Albuquerque, New Mexico 87123, United States.
J Chem Theory Comput. 2021 Nov 9;17(11):7056-7084. doi: 10.1021/acs.jctc.1c00628. Epub 2021 Oct 26.
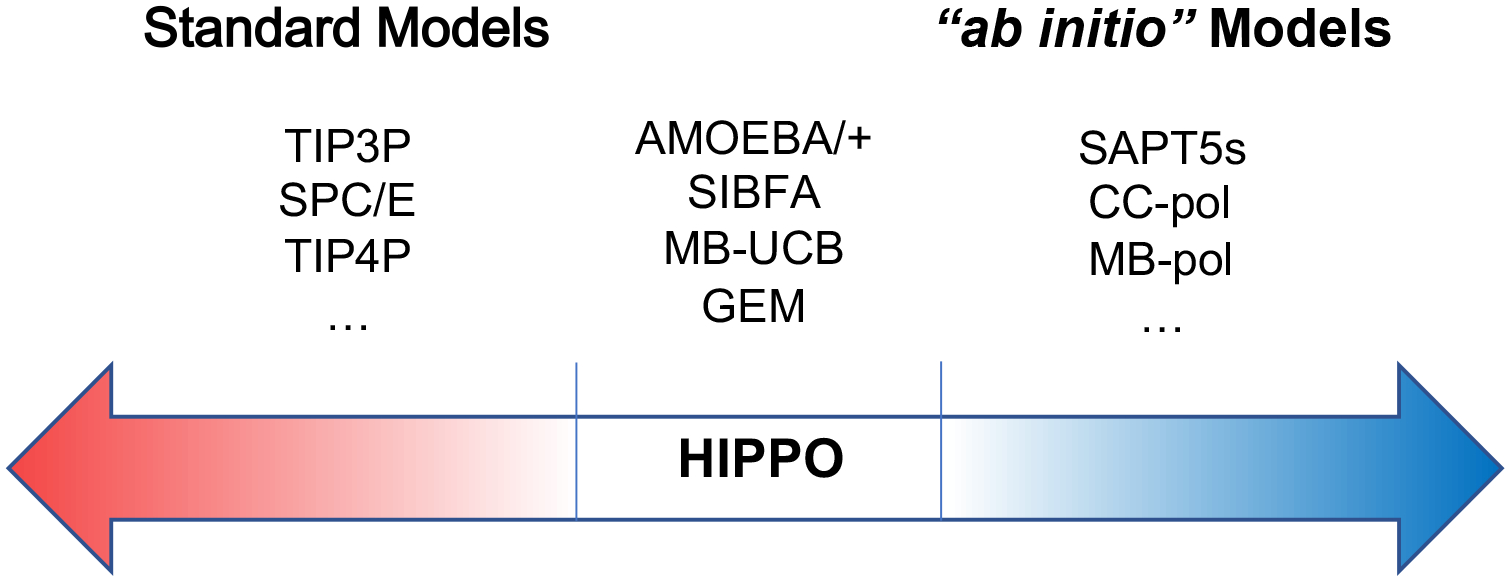
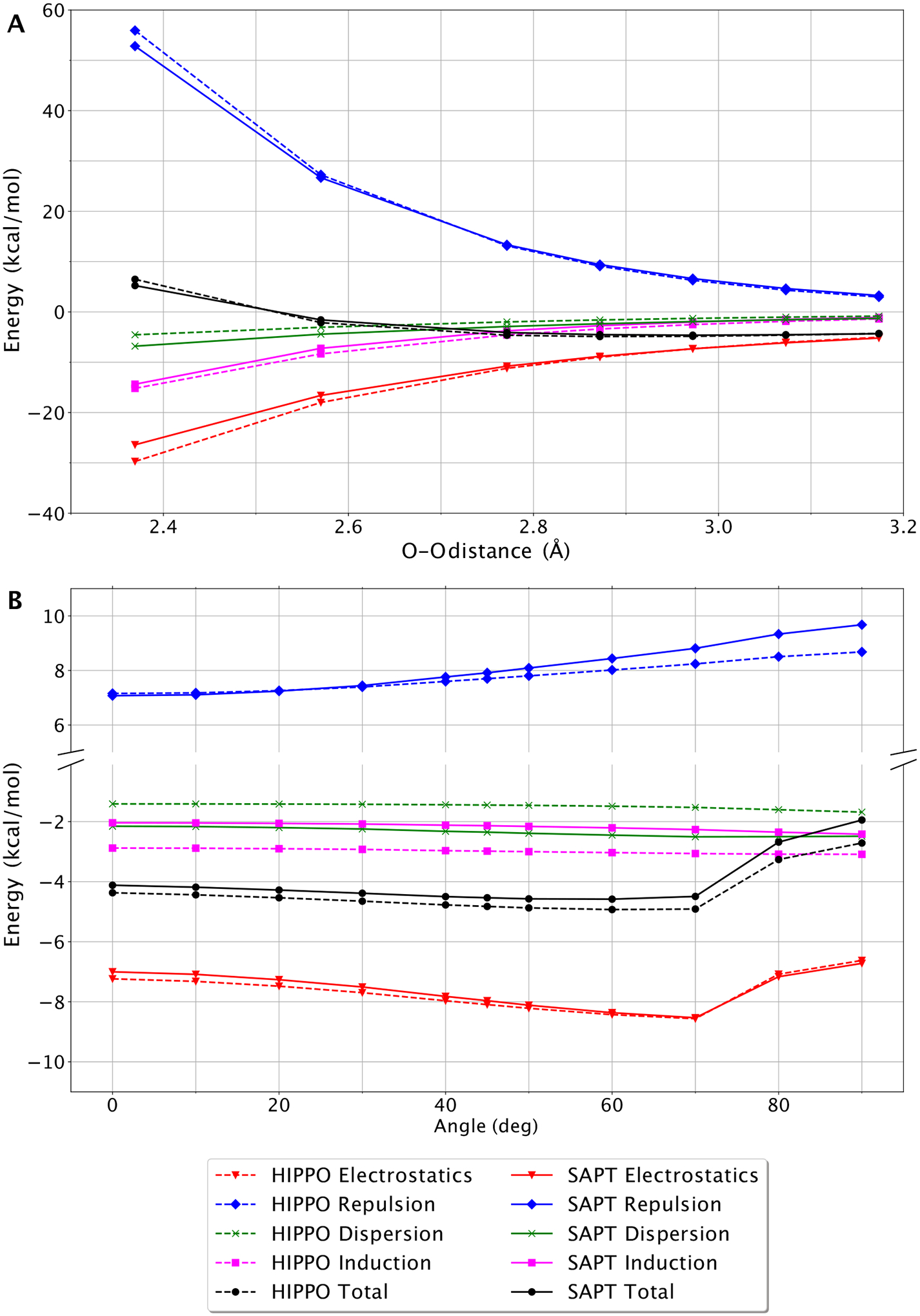
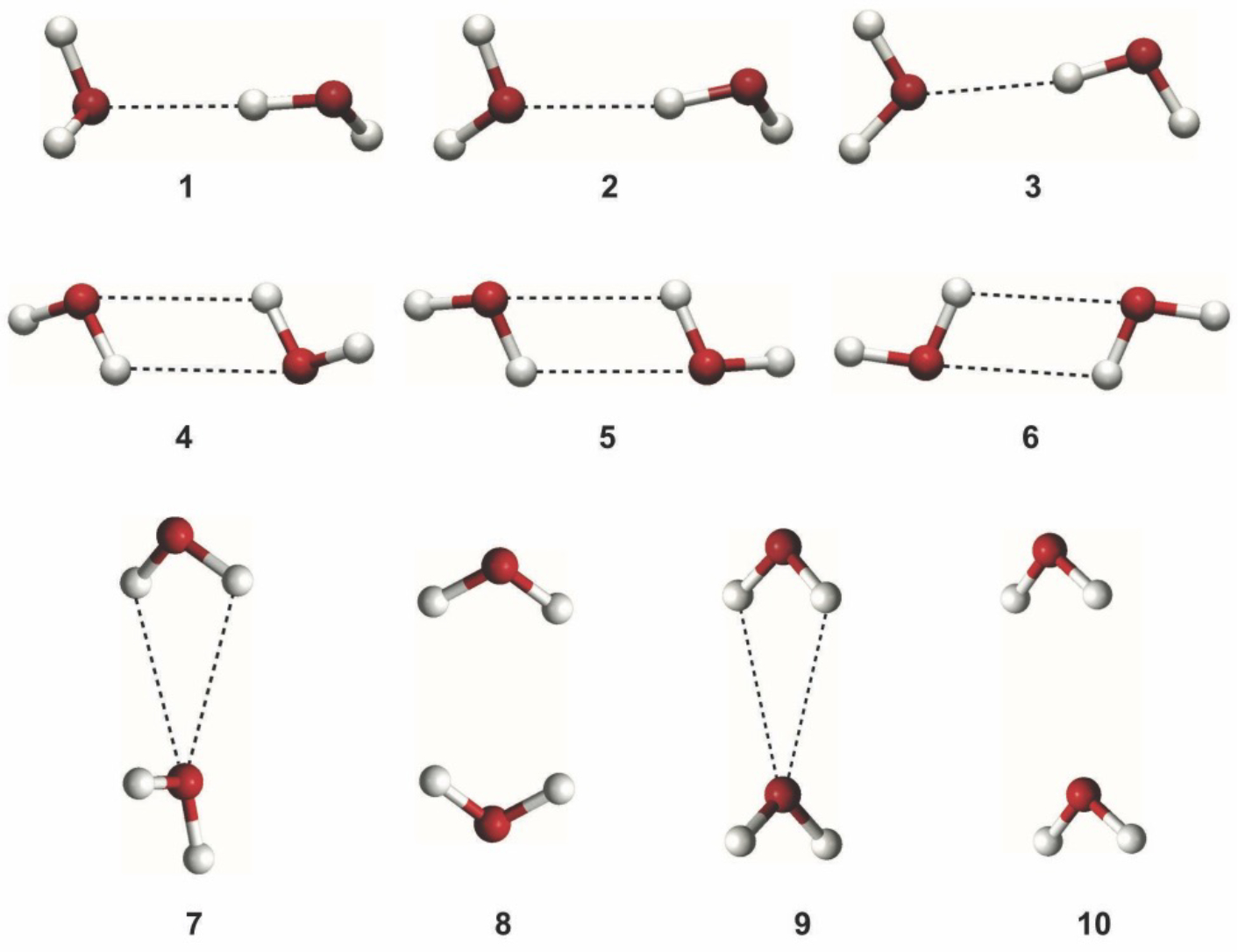
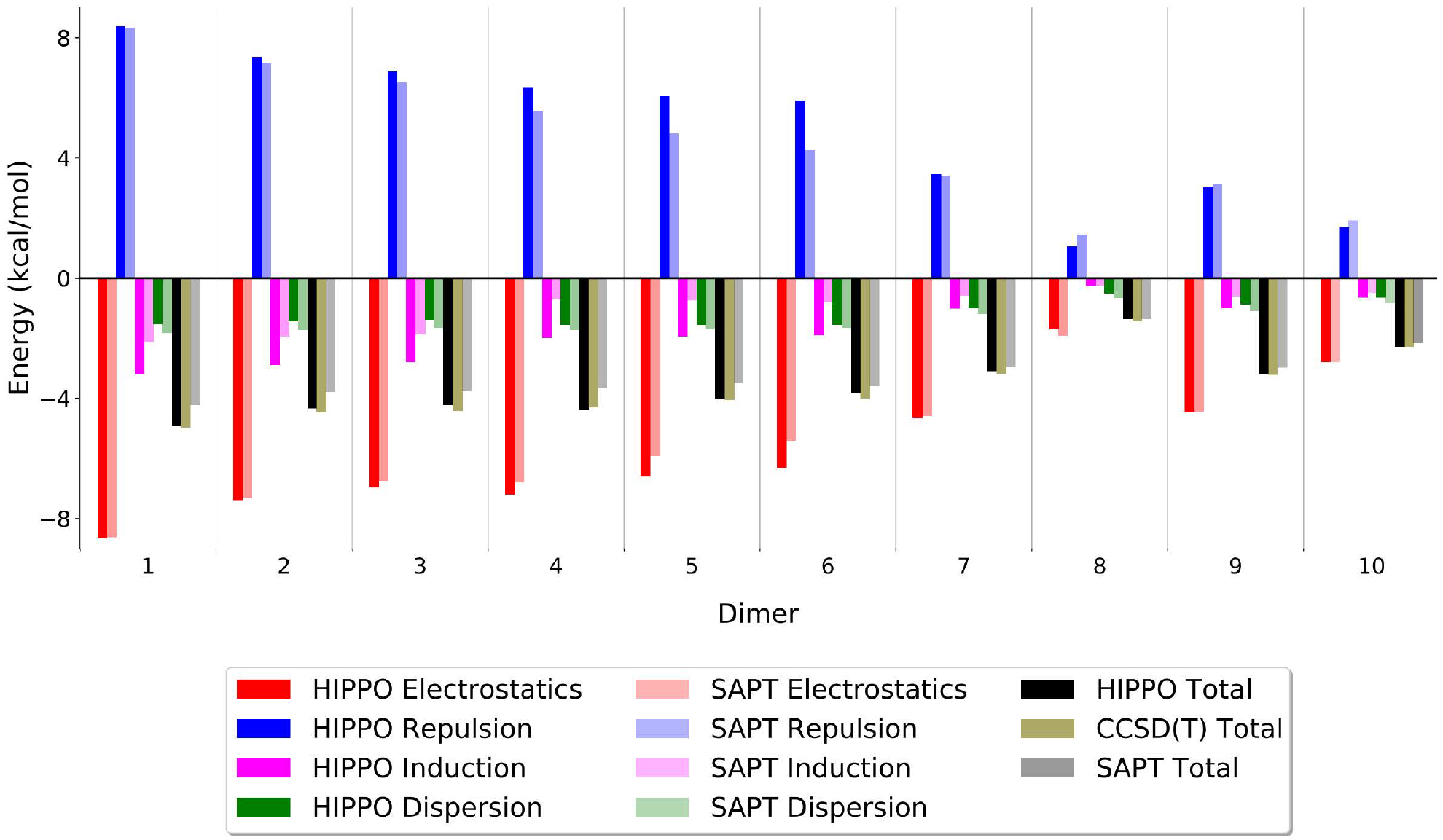
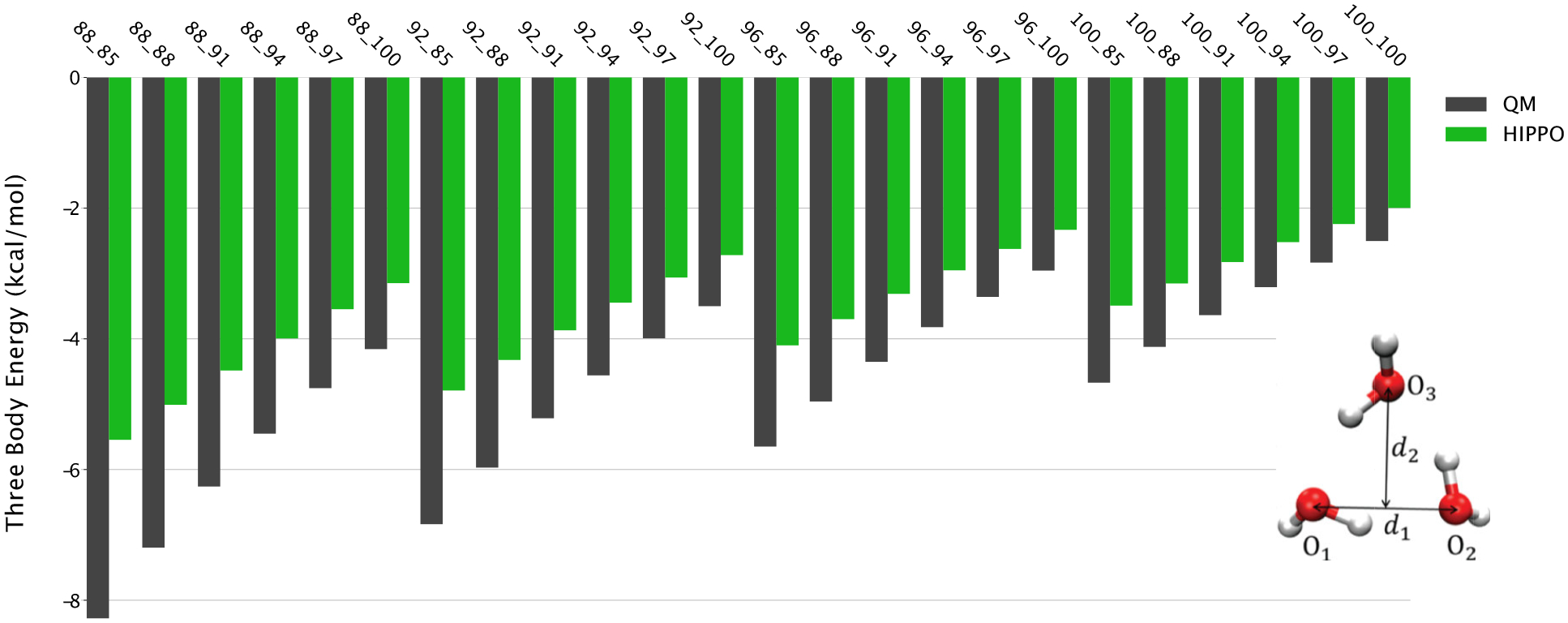
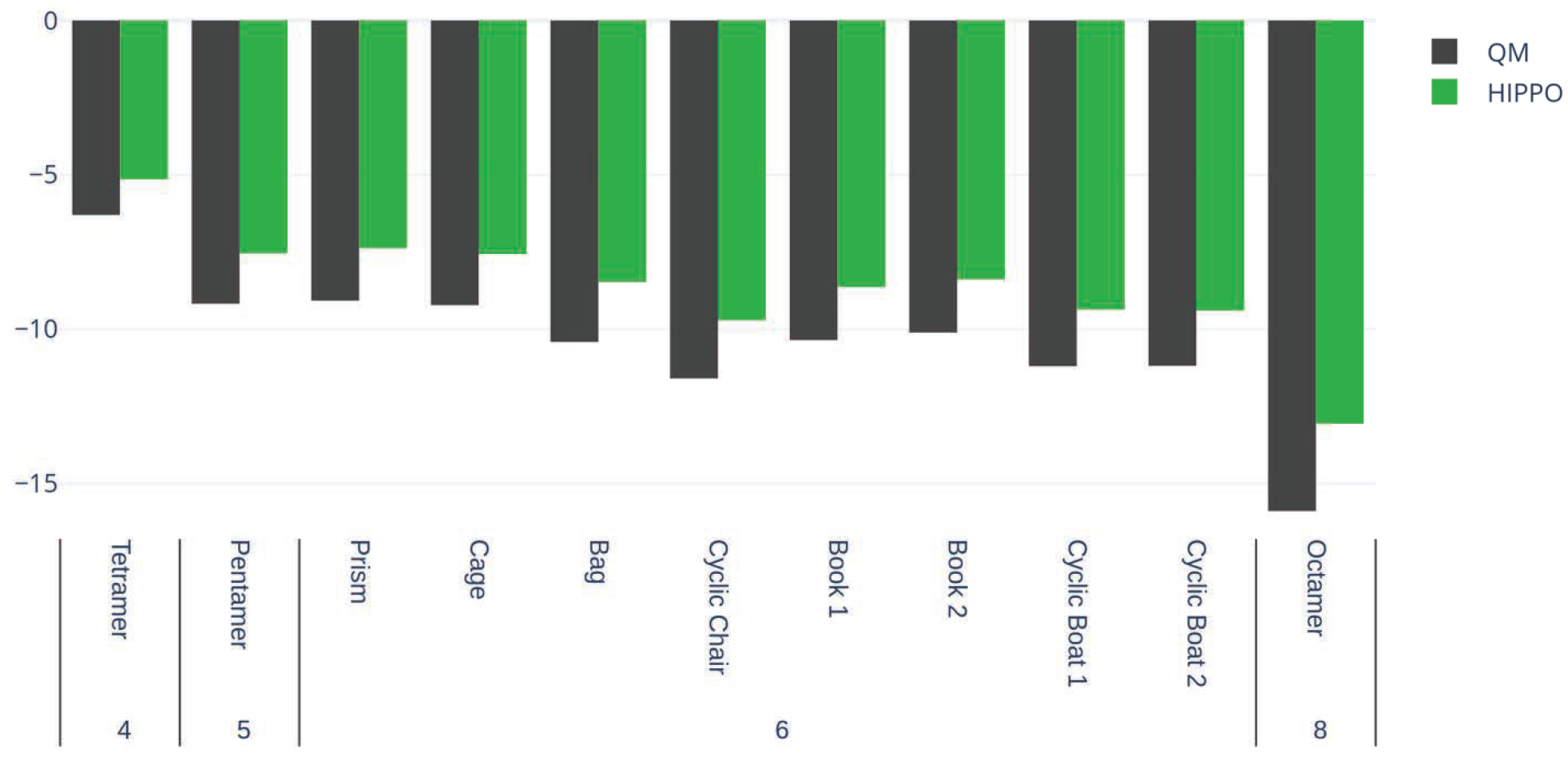
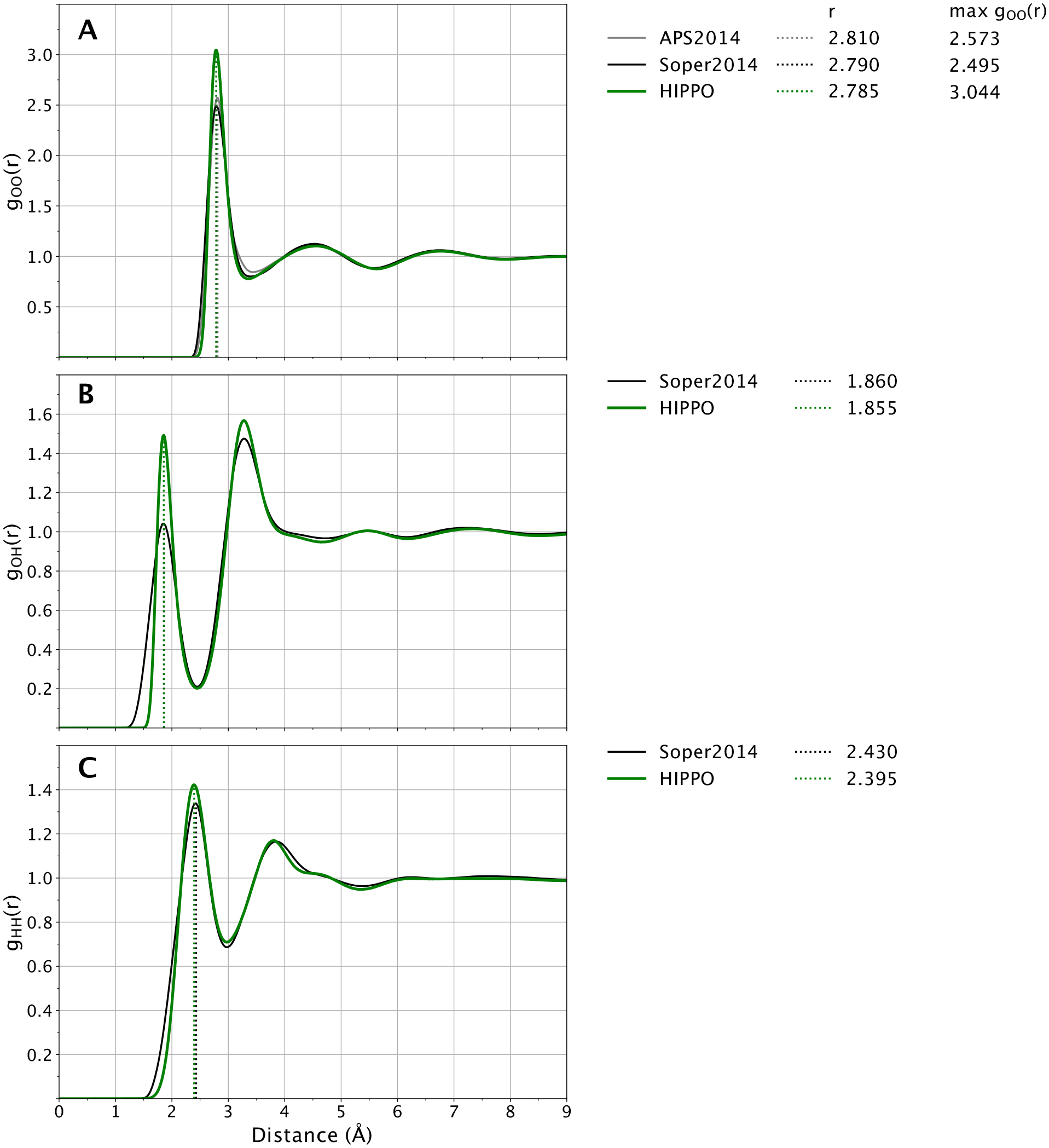
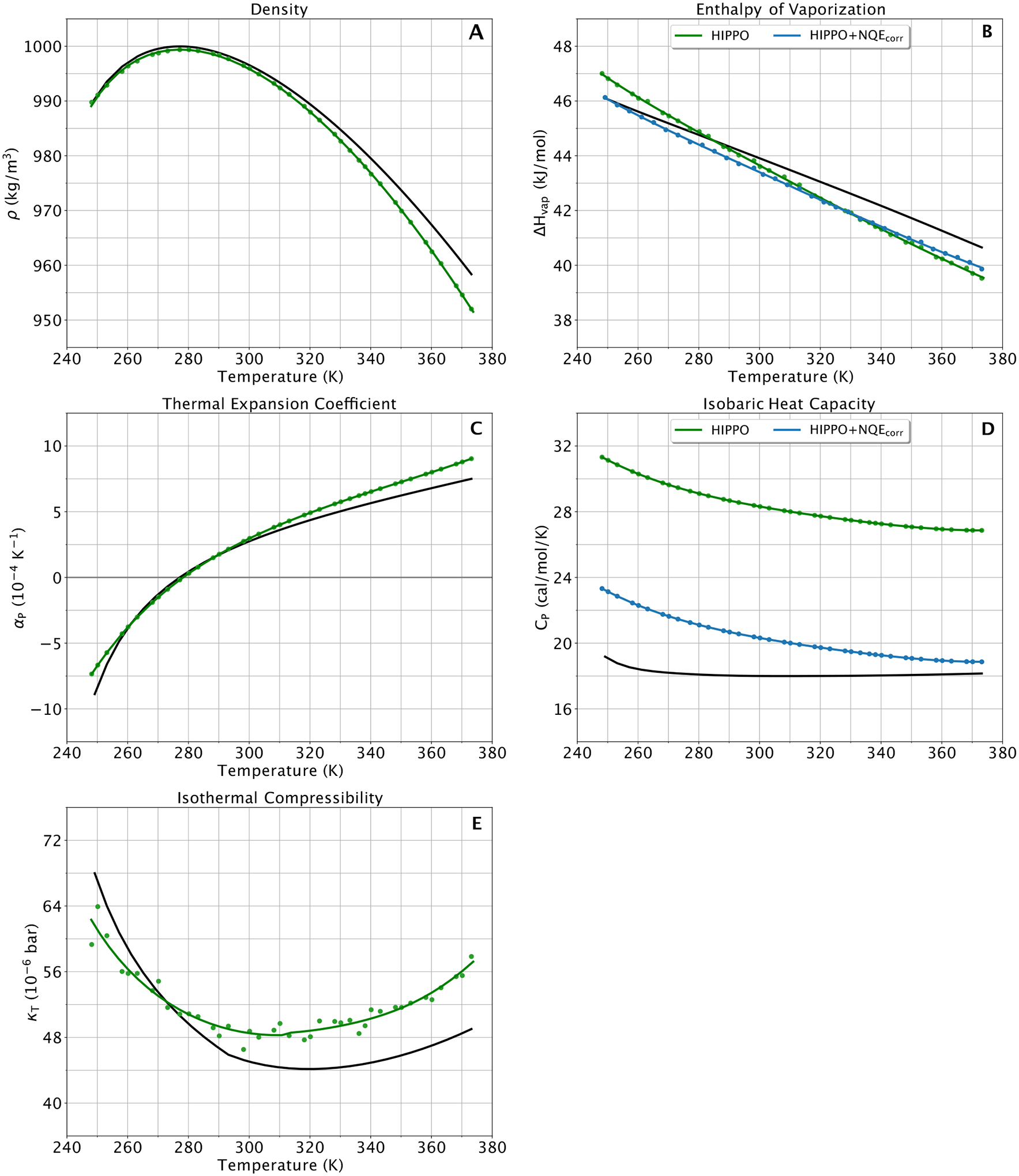
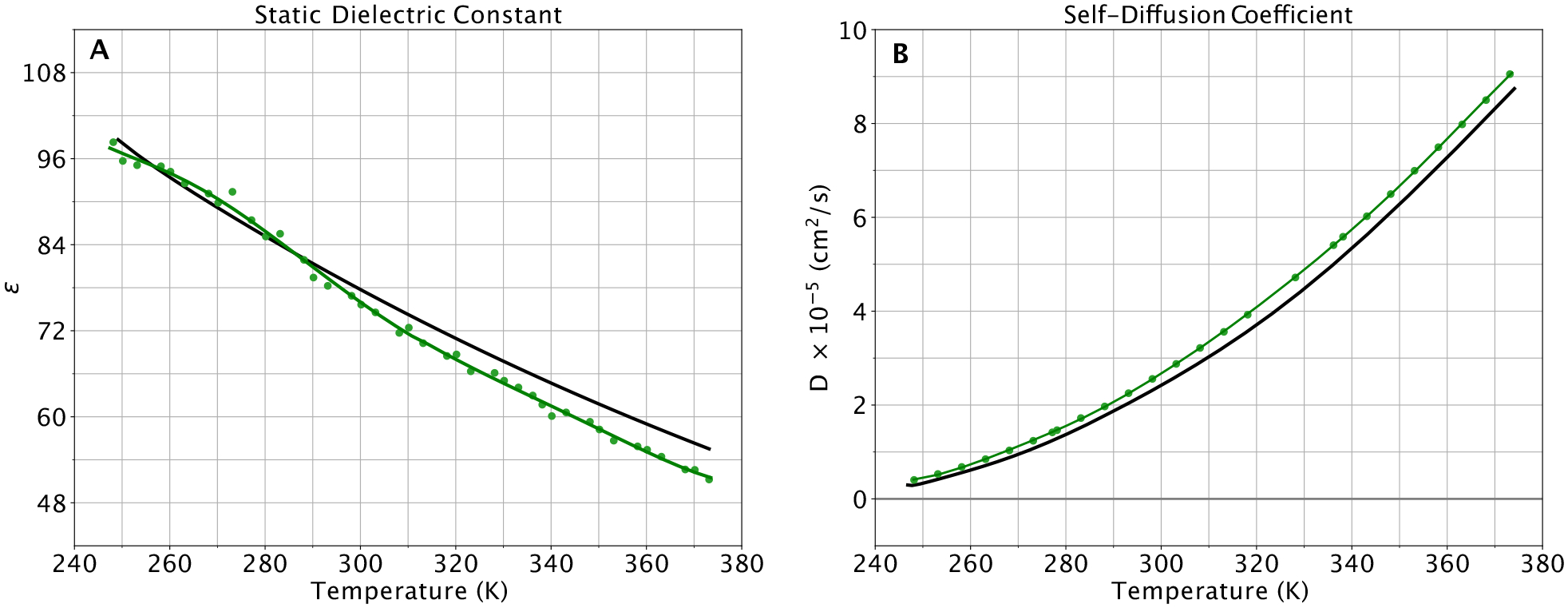
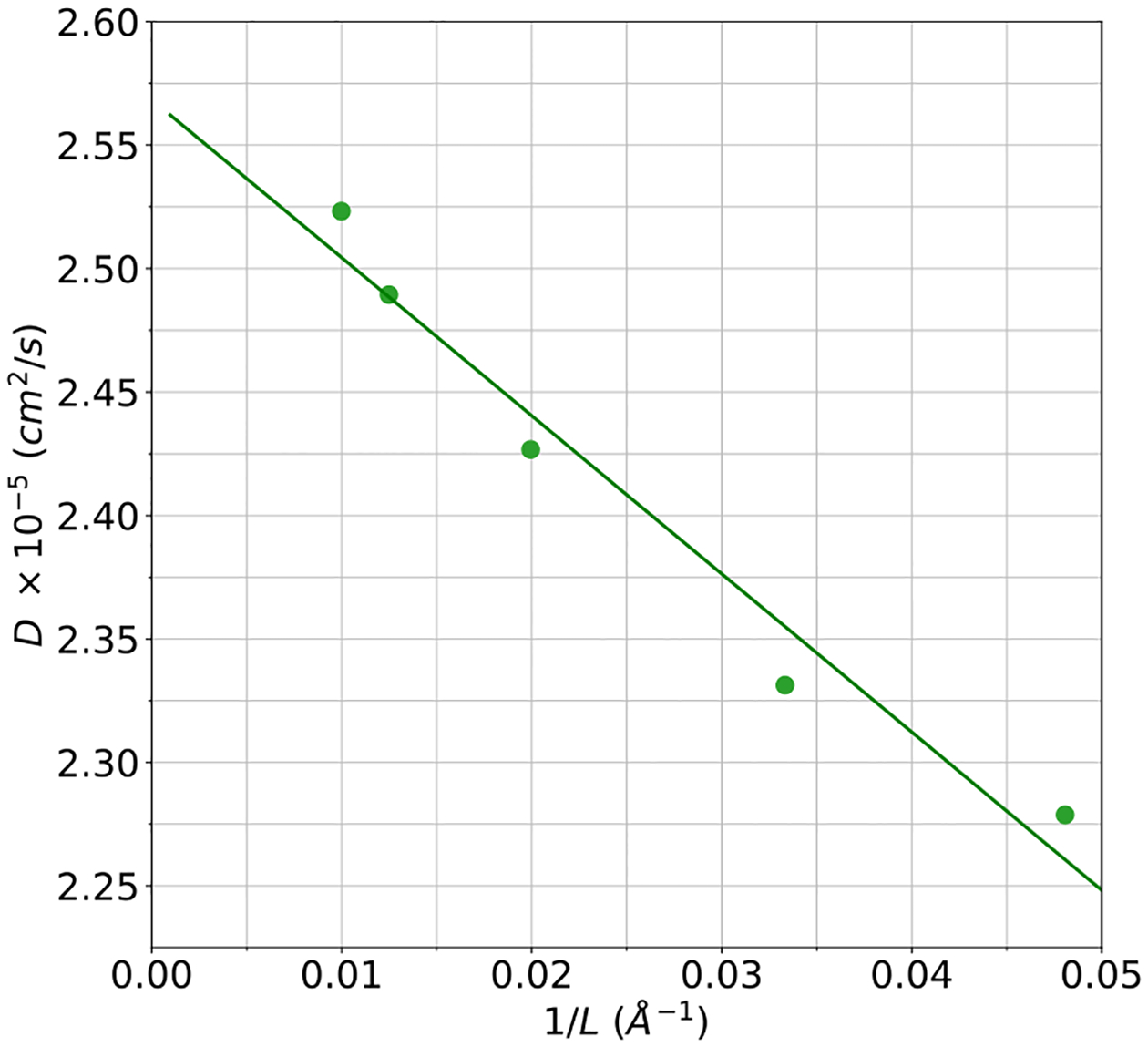
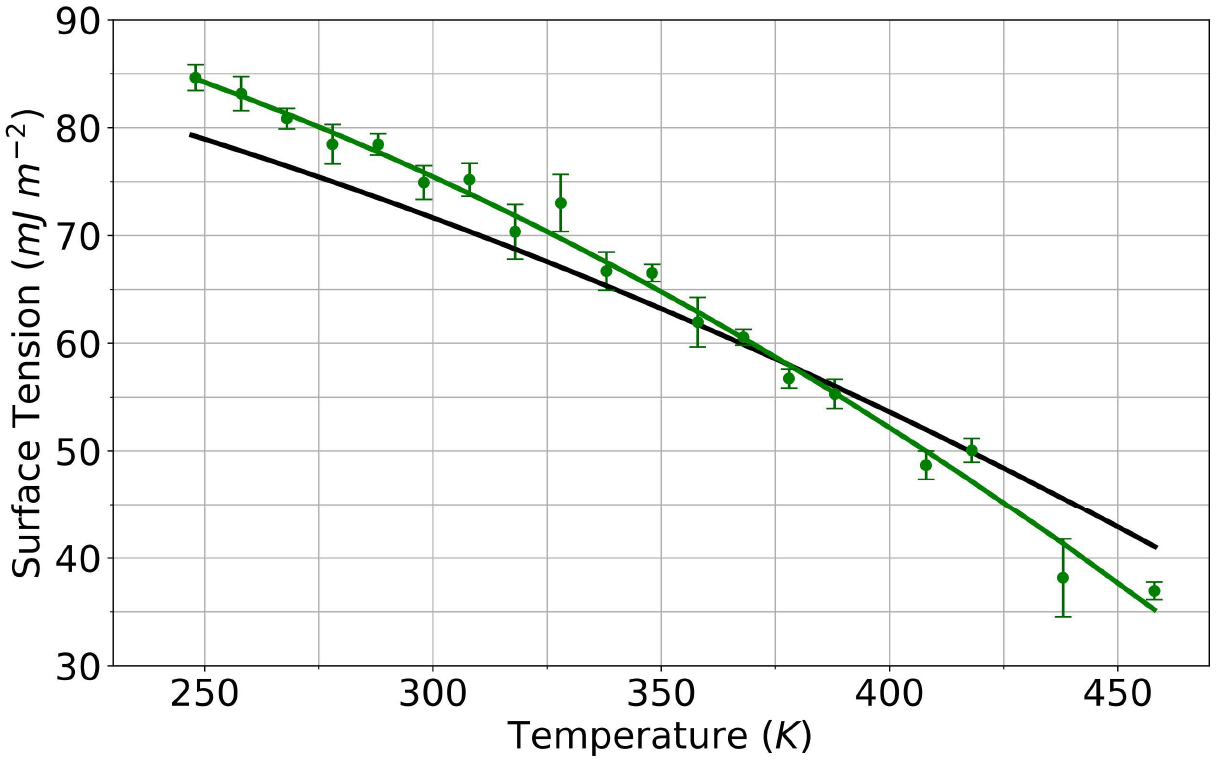
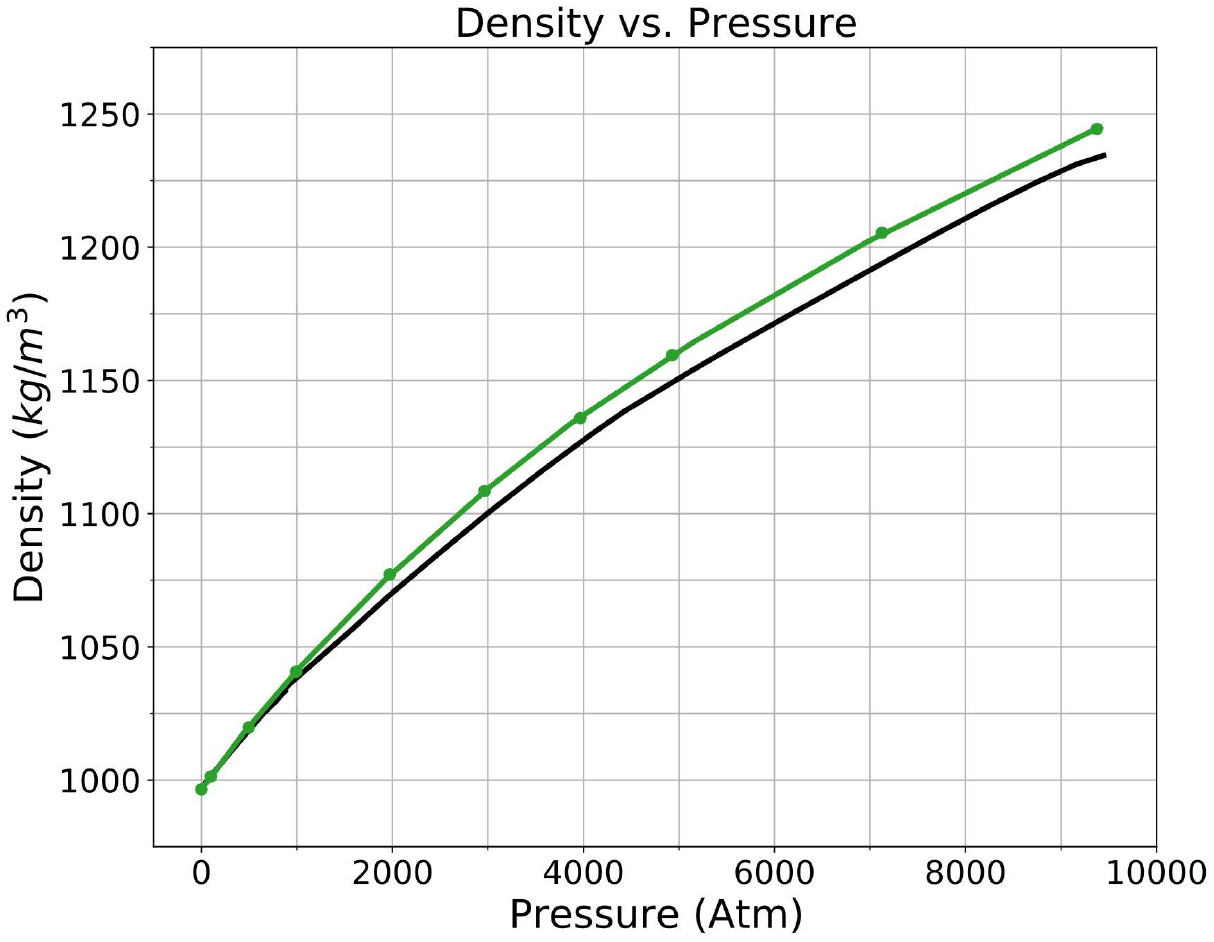
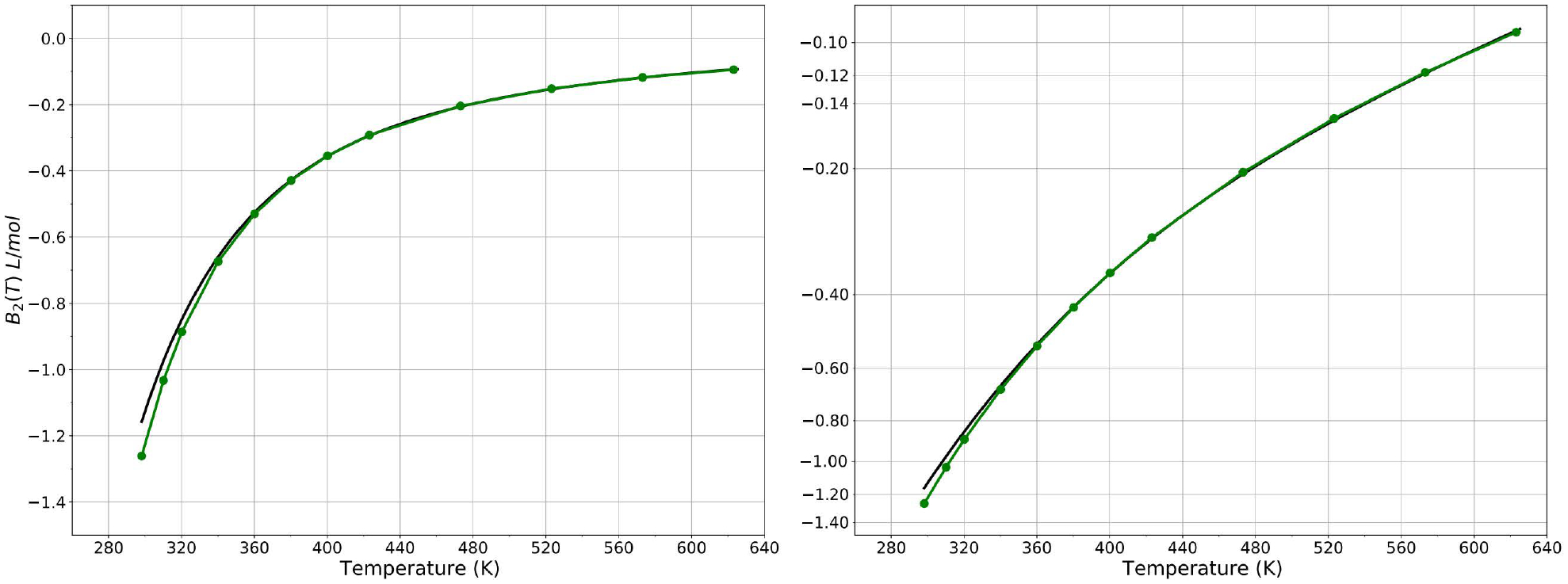
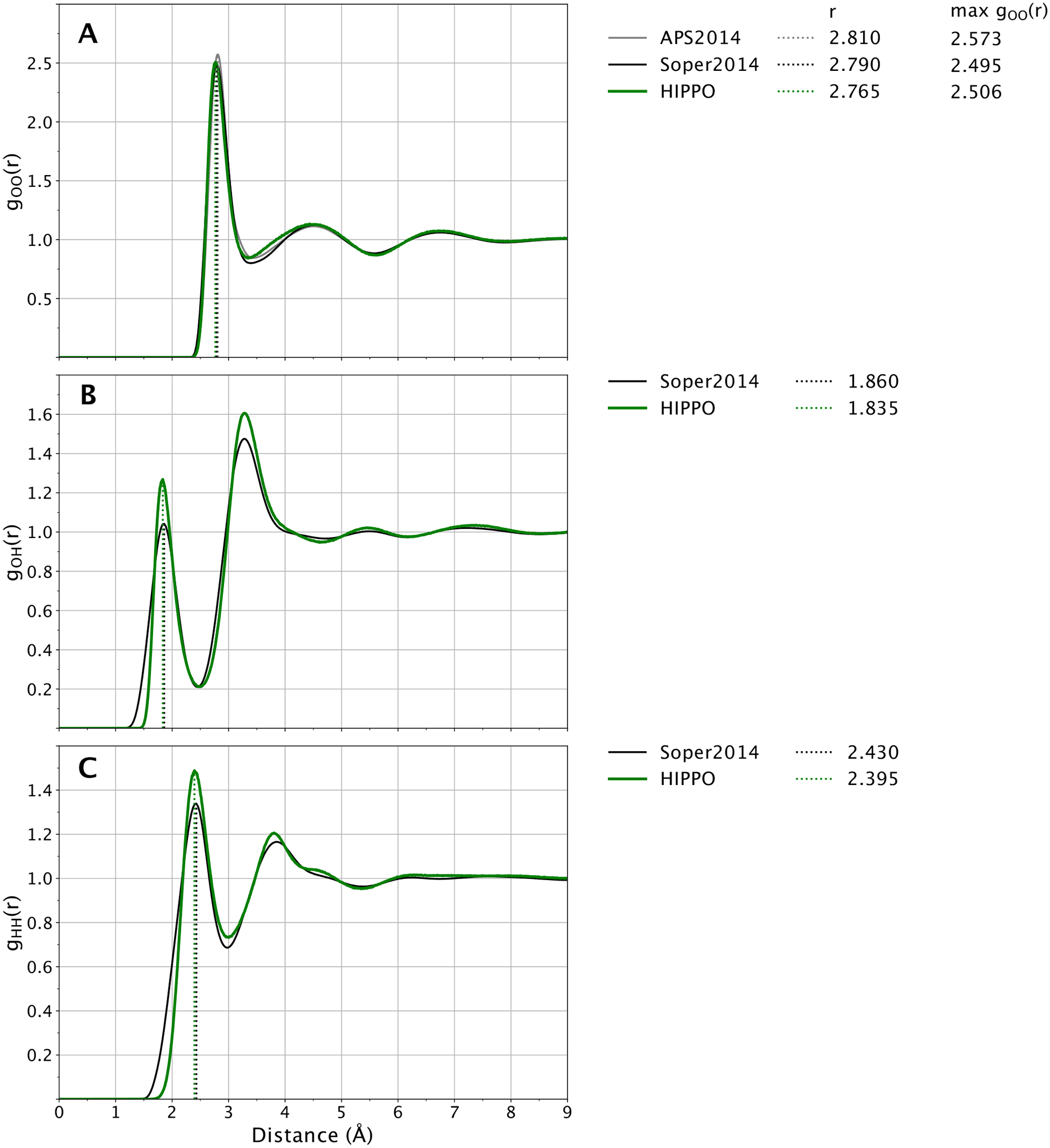
A new empirical potential for efficient, large scale molecular dynamics simulation of water is presented. The HIPPO (Hydrogen-like Intermolecular Polarizable POtential) force field is based upon the model electron density of a hydrogen-like atom. This framework is used to derive and parametrize individual terms describing charge penetration damped permanent electrostatics, damped polarization, charge transfer, anisotropic Pauli repulsion, and damped dispersion interactions. Initial parameter values were fit to Symmetry Adapted Perturbation Theory (SAPT) energy components for ten water dimer configurations, as well as the radial and angular dependence of the canonical dimer. The SAPT-based parameters were then systematically refined to extend the treatment to water bulk phases. The final HIPPO water model provides a balanced representation of a wide variety of properties of gas phase clusters, liquid water, and ice polymorphs, across a range of temperatures and pressures. This water potential yields a rationalization of water structure, dynamics, and thermodynamics explicitly correlated with an energy decomposition, while providing a level of accuracy comparable or superior to previous polarizable atomic multipole force fields. The HIPPO water model serves as a cornerstone around which similarly detailed physics-based models can be developed for additional molecular species.
提出了一种新的经验势,可有效、大规模地模拟水分子动力学。HIPPO(类氢分子极化势能)力场基于类氢原子的模型电子密度。该框架用于推导和参数化描述电荷穿透衰减永久静电、衰减极化、电荷转移、各向异性 Pauli 排斥和衰减色散相互作用的各个项。初始参数值拟合到十水二聚体构型的对称自适应微扰理论(SAPT)能量分量,以及正则二聚体的径向和角度依赖性。然后,基于 SAPT 的参数被系统地细化,以将处理扩展到水的体相。最终的 HIPPO 水模型提供了对气相团簇、液态水和冰多型体的各种性质的平衡描述,涵盖了一系列温度和压力范围。这种水势明确地与能量分解相关联,对水结构、动力学和热力学进行合理化,同时提供了与以前的极化原子多极力场相当或更高的精度水平。HIPPO 水模型是一个基石,可以围绕它开发其他分子物种的类似详细的物理模型。