Broad Institute of MIT and Harvard, Cambridge, MA, USA.
Genentech, South San Francisco, CA, USA.
Nat Methods. 2021 Nov;18(11):1352-1362. doi: 10.1038/s41592-021-01264-7. Epub 2021 Oct 28.
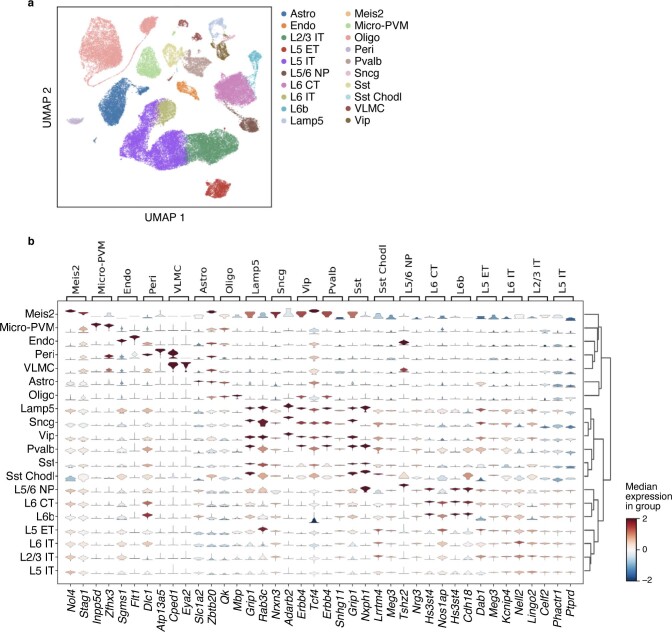
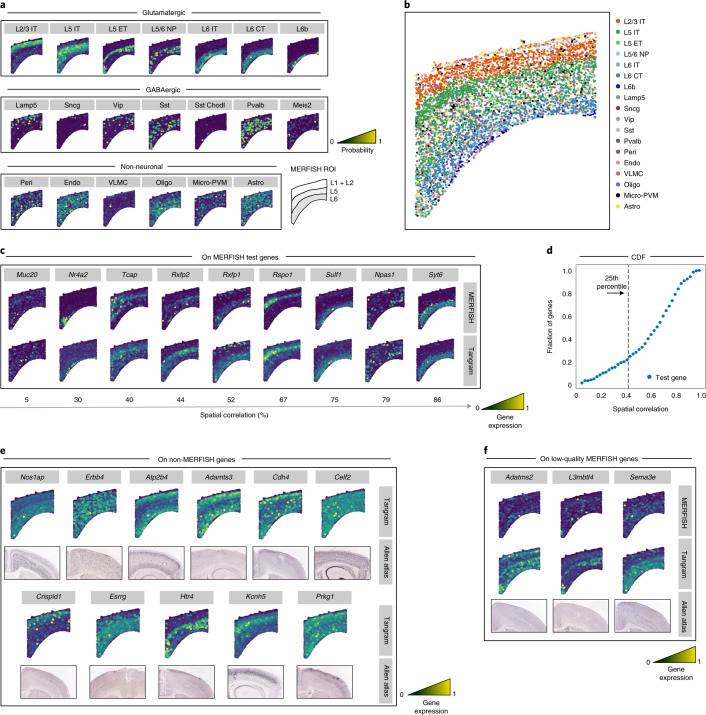
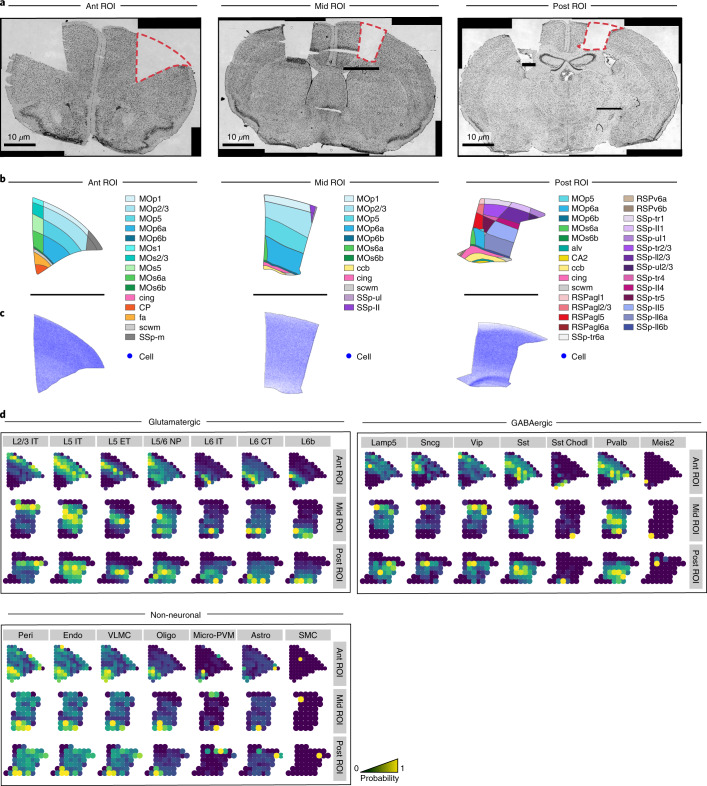
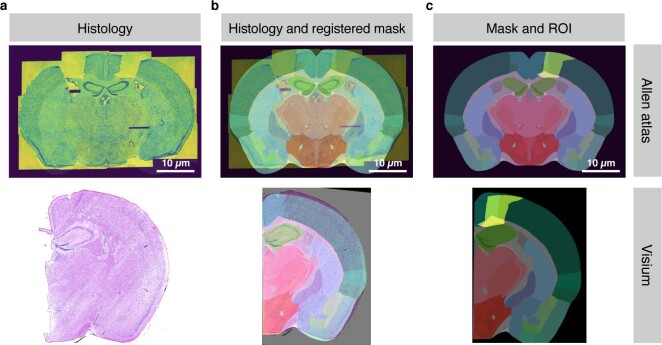
Charting an organs' biological atlas requires us to spatially resolve the entire single-cell transcriptome, and to relate such cellular features to the anatomical scale. Single-cell and single-nucleus RNA-seq (sc/snRNA-seq) can profile cells comprehensively, but lose spatial information. Spatial transcriptomics allows for spatial measurements, but at lower resolution and with limited sensitivity. Targeted in situ technologies solve both issues, but are limited in gene throughput. To overcome these limitations we present Tangram, a method that aligns sc/snRNA-seq data to various forms of spatial data collected from the same region, including MERFISH, STARmap, smFISH, Spatial Transcriptomics (Visium) and histological images. Tangram can map any type of sc/snRNA-seq data, including multimodal data such as those from SHARE-seq, which we used to reveal spatial patterns of chromatin accessibility. We demonstrate Tangram on healthy mouse brain tissue, by reconstructing a genome-wide anatomically integrated spatial map at single-cell resolution of the visual and somatomotor areas.
绘制器官的生物图谱需要我们在空间上解析整个单细胞转录组,并将这些细胞特征与解剖学尺度联系起来。单细胞和单核 RNA 测序 (sc/snRNA-seq) 可以全面分析细胞,但会丢失空间信息。空间转录组学允许进行空间测量,但分辨率较低,灵敏度有限。靶向原位技术解决了这两个问题,但基因通量有限。为了克服这些限制,我们提出了 Tangram 方法,该方法可以将 sc/snRNA-seq 数据与从同一区域收集的各种形式的空间数据进行对齐,包括 MERFISH、STARmap、smFISH、Spatial Transcriptomics (Visium) 和组织学图像。Tangram 可以映射任何类型的 sc/snRNA-seq 数据,包括来自 SHARE-seq 的多模态数据,我们使用它来揭示染色质可及性的空间模式。我们在健康的小鼠脑组织上展示了 Tangram,通过重建视觉和躯体运动区域的全基因组解剖学整合的单细胞分辨率的空间图谱来实现。