Liang Guangcai
School of Chemical Engineering and Technology, Tianjin University, Yaguan Road 135, Jinnan District, Tianjin, 300350, China.
Synth Syst Biotechnol. 2021 Nov 11;6(4):377-383. doi: 10.1016/j.synbio.2021.10.003. eCollection 2021 Dec.
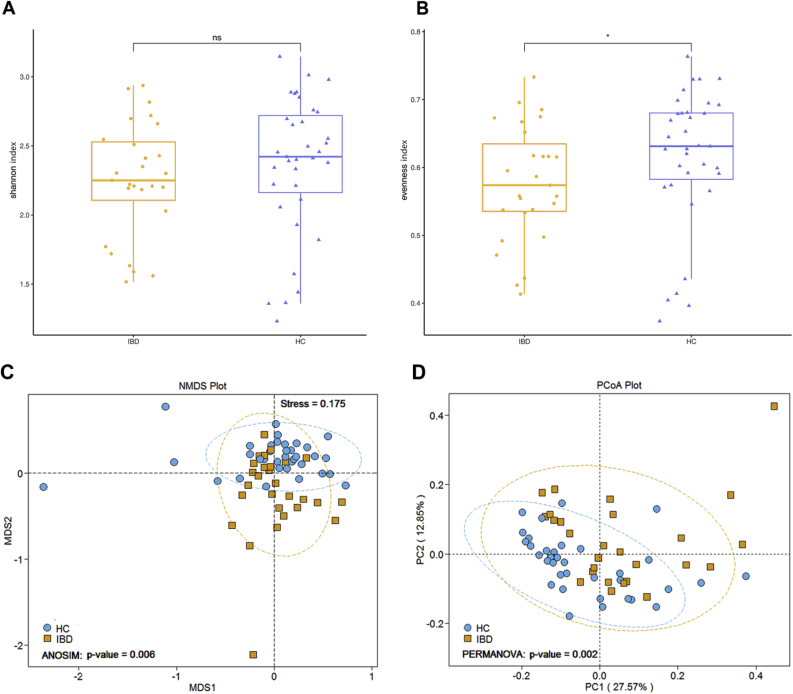
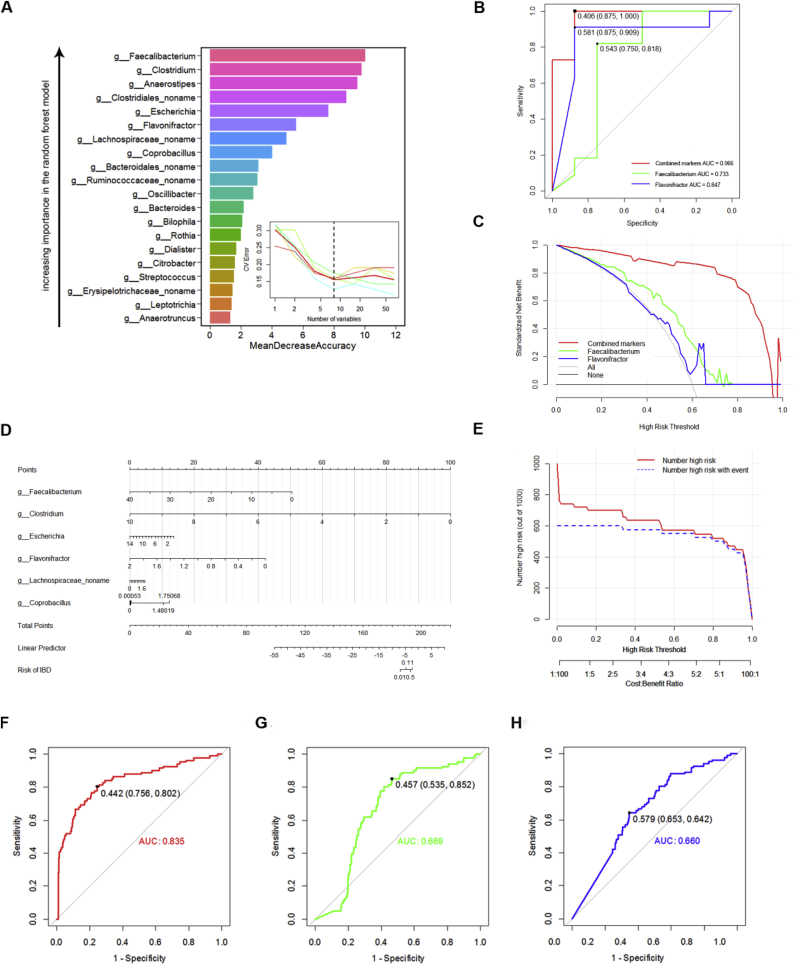
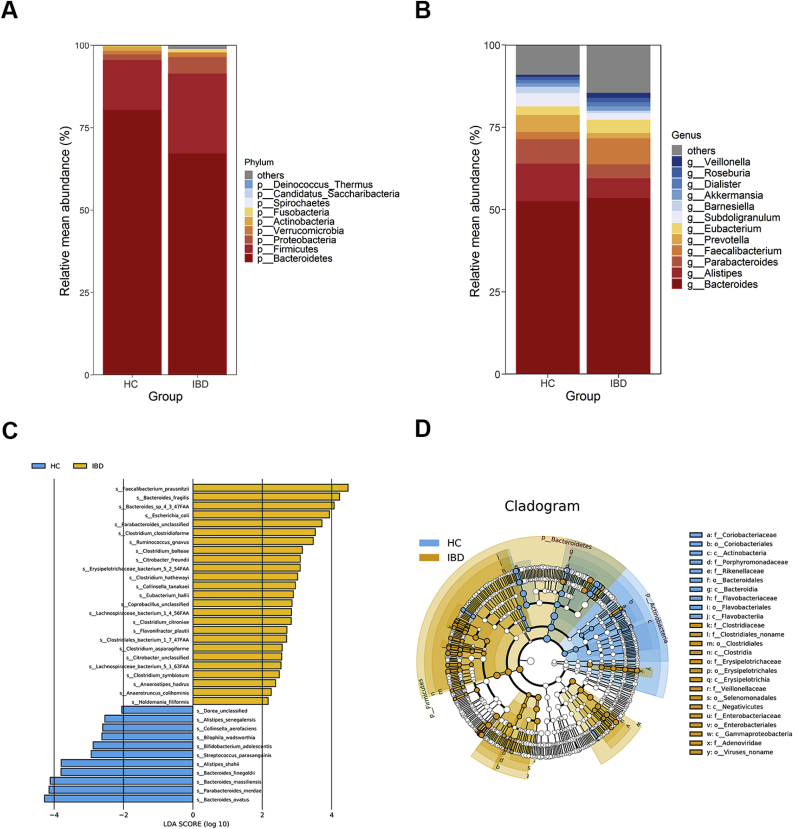
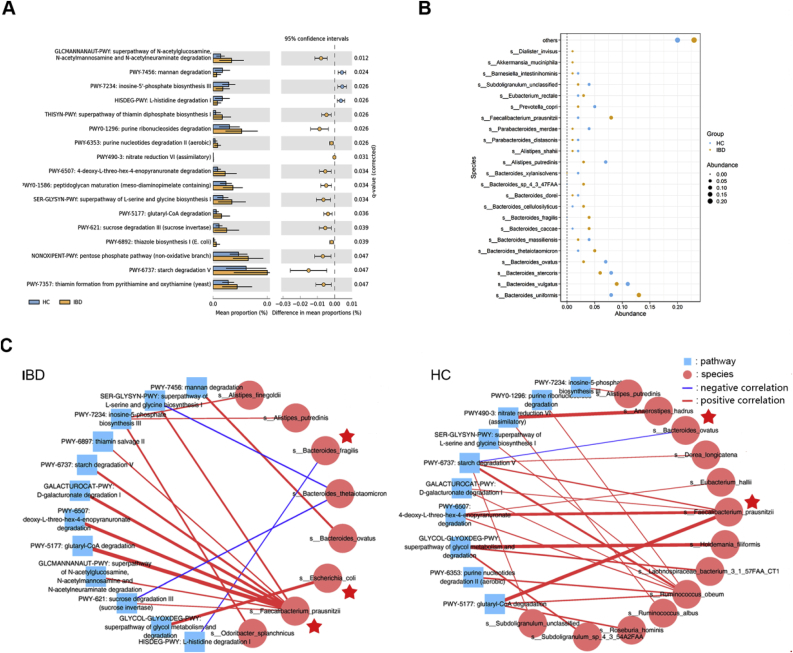
Dysregulation of the gut microbiome has been implicated in the progression of many diseases. This study explored the role of microbial and metabolic signatures, and their interaction between the Human inflammatory bowel disease (IBD) and healthy controls (HCs) based on the combination of machine learning and traditional statistical analysis, using data collected from the Human Microbiome Project (HMP) and the Integrative Human Microbiome Project (iHMP). It was showed that the microbial and metabolic signatures of IBD patients were significantly different from those of HCs. Compared to HCs, IBD subjects were characterized by 25 enriched species and 6 depleted species. Furthermore, a total of 17 discriminative pathways were identified between the IBD and HC groups. Those differential pathways were mainly involved in amino acid, nucleotide biosynthesis, and carbohydrate degradation. Notably, co-occurrence network analysis revealed that non-predominant bacteria and predominant bacteria formed the same broad and strong co-occurring relationships with pathways. Moreover, the essay identified a combinatorial marker panel that could distinguish IBD from HCs. Receiver Operating Characteristic (ROC) and Decision Curve Analysis (DCA) confirmed the high accuracy (AUC = 0.966) and effectiveness of the model. Meanwhile, an independent cohort used for external validation also showed the identical high efficacy (AUC = 0.835). These findings showed that the gut microbes may be relevant to the pathogenesis and pathophysiology, and offer universal utility as a non-invasive diagnostic test in IBD.
肠道微生物群失调与多种疾病的进展有关。本研究基于机器学习和传统统计分析相结合的方法,利用从人类微生物组计划(HMP)和综合人类微生物组计划(iHMP)收集的数据,探讨了微生物和代谢特征在人类炎症性肠病(IBD)与健康对照(HCs)之间的作用及其相互作用。结果表明,IBD患者的微生物和代谢特征与HCs显著不同。与HCs相比,IBD受试者的特征是有25种富集物种和6种减少物种。此外,在IBD组和HC组之间共鉴定出17条有鉴别意义的代谢途径。这些差异途径主要涉及氨基酸、核苷酸生物合成和碳水化合物降解。值得注意的是,共现网络分析表明,非优势菌和优势菌与代谢途径形成了相同广泛而强烈的共现关系。此外,本文还确定了一个可以区分IBD和HCs的组合标志物面板。受试者工作特征(ROC)和决策曲线分析(DCA)证实了该模型的高准确性(AUC = 0.966)和有效性。同时,用于外部验证的独立队列也显示出相同的高疗效(AUC = 0.835)。这些发现表明,肠道微生物可能与发病机制和病理生理学相关,并作为IBD的一种非侵入性诊断测试具有广泛的实用性。