Xu Xinwei, Ocansey Dickson Kofi Wiredu, Hang Sanhua, Wang Bo, Amoah Samuel, Yi Chengxue, Zhang Xu, Liu Lianqin, Mao Fei
Key Laboratory of Medical Science and Laboratory Medicine of Jiangsu Province, School of Medicine, Jiangsu University, Zhenjiang, 212013, Jiangsu, People's Republic of China.
Directorate of University Health Services, University of Cape Coast, PMB, Cape Coast, Ghana.
Gut Pathog. 2022 Jun 21;14(1):26. doi: 10.1186/s13099-022-00499-9.
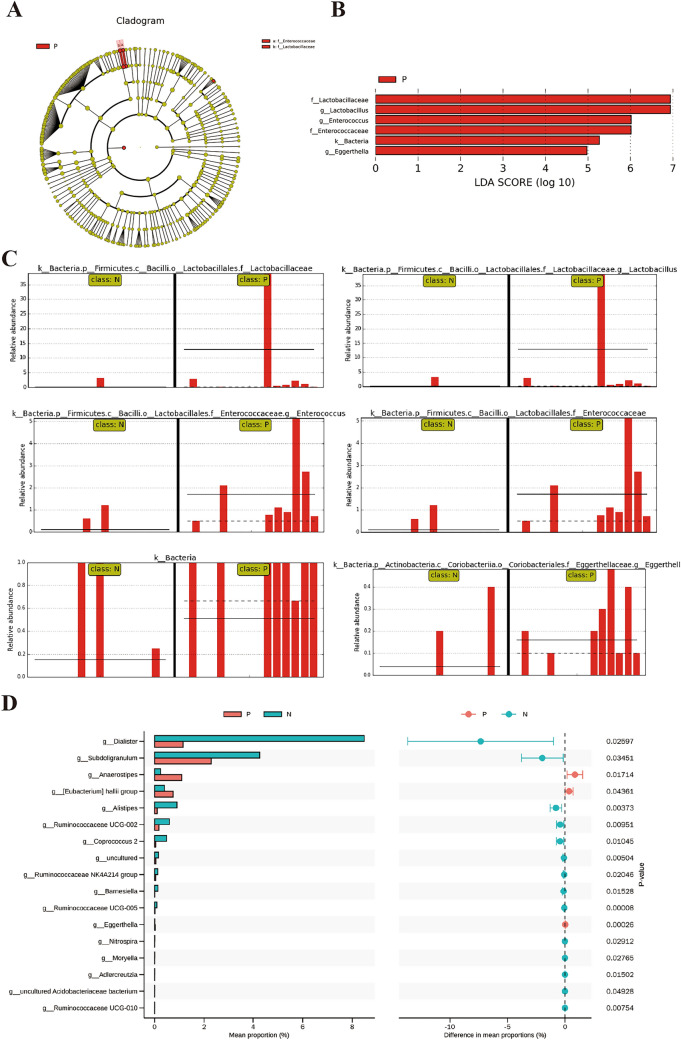
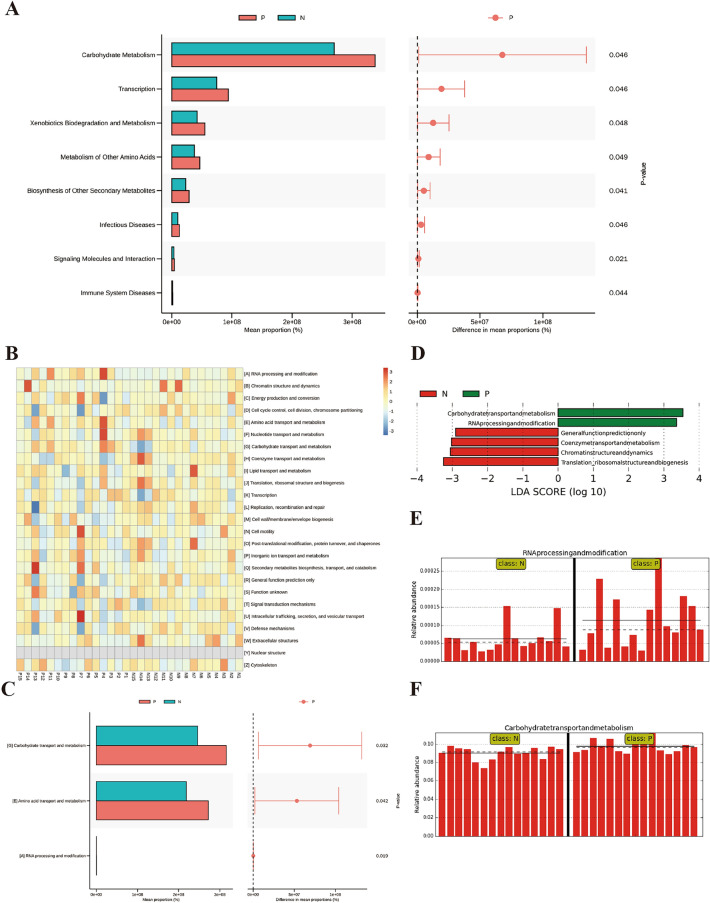
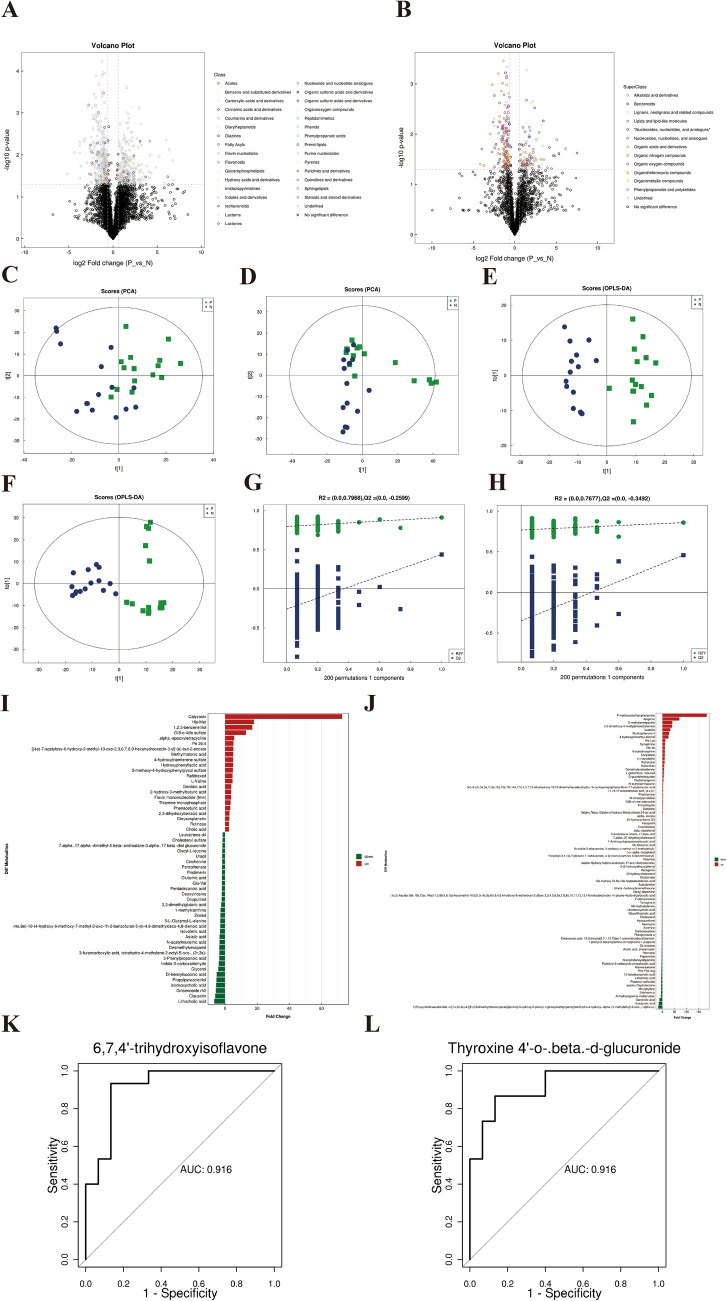
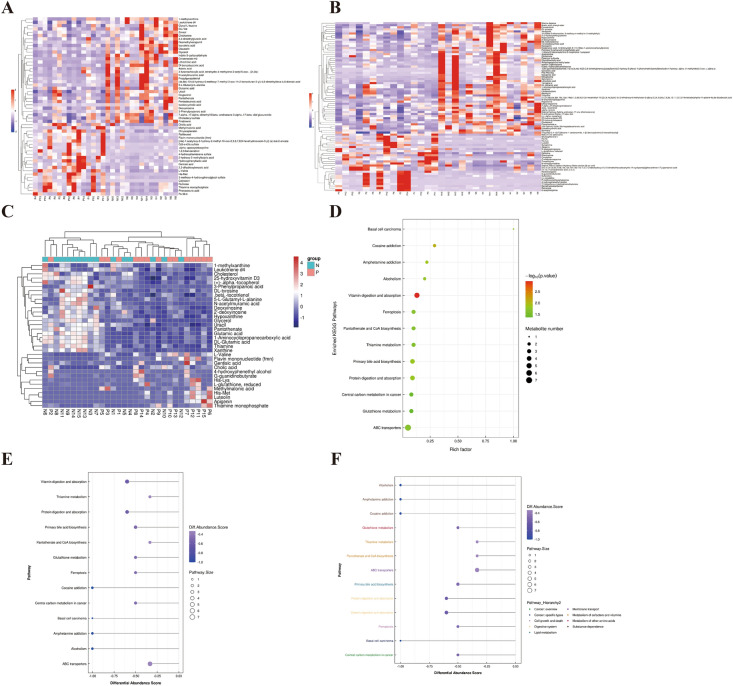
Inflammatory bowel disease (IBD), a chronic gut immune dysregulation and dysbiosis condition is rapidly increasing in global incidence. Regardless, there is a lack of ideal diagnostic markers, while conventional treatment provides scarce desired results, thus, the exploration for better options. Changes in the gut microbial composition and metabolites either lead to or are caused by the immune dysregulation that characterizes IBD. This study examined the fecal metagenomics and metabolomic changes in IBD patients. A total of 30 fecal samples were collected from 15 IBD patients and 15 healthy controls for 16S rDNA gene sequencing and UHPLC/Q-TOF-MS detection of metabolomics. Results showed that there was a severe perturbation of gut bacteria community composition, diversity, metabolites, and associated functions and metabolic pathways in IBD. This included a significantly decreased abundance of Bacteroidetes and Firmicutes, increased disease-associated phyla such as Proteobacteria and Actinobacteria, and increased Escherichia coli and Klebsiella pneumoniae in IBD. A total of 3146 metabolites were detected out of which 135 were differentially expressed between IBD and controls. Metabolites with high sensitivity and specificity in differentiating IBD from healthy individuals included 6,7,4'-trihydroxyisoflavone and thyroxine 4'-o-.beta.-d-glucuronide (AUC = 0.92), normorphine and salvinorin a (AUC = 0.90), and trichostachine (AUC = 0.91). Moreover, the IBD group had significantly affected pathways including primary bile acid biosynthesis, vitamin digestion and absorption, and carbohydrate metabolism. This study reveals that the combined evaluation of metabolites and fecal microbiome can be useful to discriminate between healthy subjects and IBD patients and consequently serve as therapeutic and diagnostic targets.
炎症性肠病(IBD)是一种慢性肠道免疫失调和微生物群失调疾病,其全球发病率正在迅速上升。尽管如此,目前仍缺乏理想的诊断标志物,而传统治疗效果不佳,因此需要探索更好的治疗方案。肠道微生物组成和代谢物的变化要么导致IBD的免疫失调,要么由其引起。本研究检测了IBD患者的粪便宏基因组学和代谢组学变化。共收集了15例IBD患者和15例健康对照的30份粪便样本,进行16S rDNA基因测序和代谢组学的超高效液相色谱/四极杆飞行时间质谱检测。结果显示,IBD患者的肠道细菌群落组成、多样性、代谢物以及相关功能和代谢途径存在严重紊乱。这包括拟杆菌门和厚壁菌门的丰度显著降低,变形菌门和放线菌门等与疾病相关的菌门增加,以及IBD患者中大肠杆菌和肺炎克雷伯菌增多。共检测到3146种代谢物,其中135种在IBD患者和对照组之间差异表达。在区分IBD患者和健康个体方面具有高灵敏度和特异性的代谢物包括6,7,4'-三羟基异黄酮和甲状腺素4'-O-β-D-葡萄糖醛酸(AUC = 0.92)、去甲吗啡和鼠尾草酚(AUC = 0.90)以及知母皂苷元(AUC = 0.91)。此外,IBD组的主要胆汁酸生物合成、维生素消化和吸收以及碳水化合物代谢等途径受到显著影响。本研究表明,代谢物和粪便微生物组的联合评估有助于区分健康受试者和IBD患者,从而作为治疗和诊断靶点。