U.S. Department of Energy, Joint Genome Institute, Berkeley, CA, USA.
Environmental Genomics and Systems Biology Division, Lawrence Berkeley National Laboratory, Berkeley, CA, USA.
ISME J. 2022 May;16(5):1337-1347. doi: 10.1038/s41396-021-01178-4. Epub 2021 Dec 30.
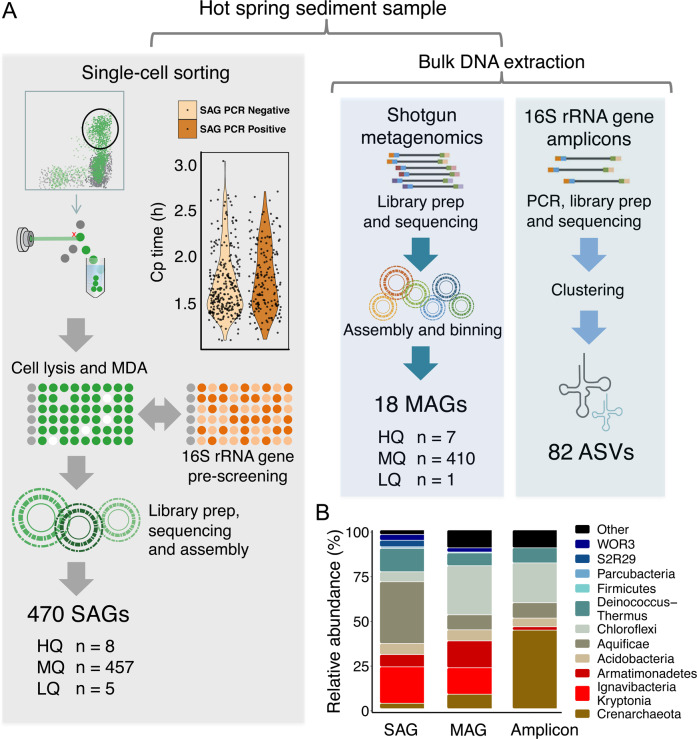
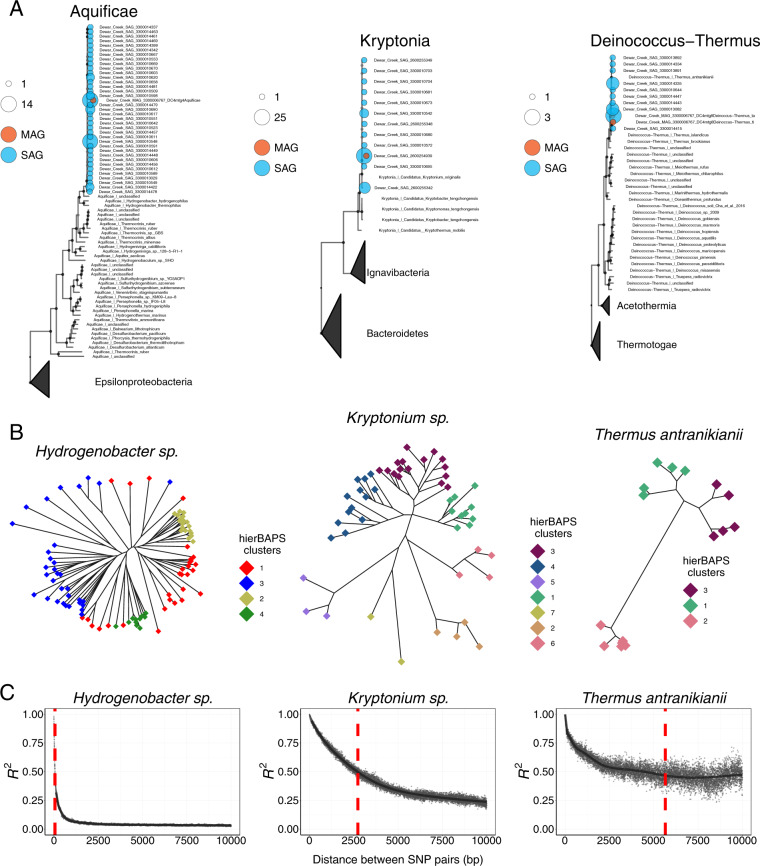
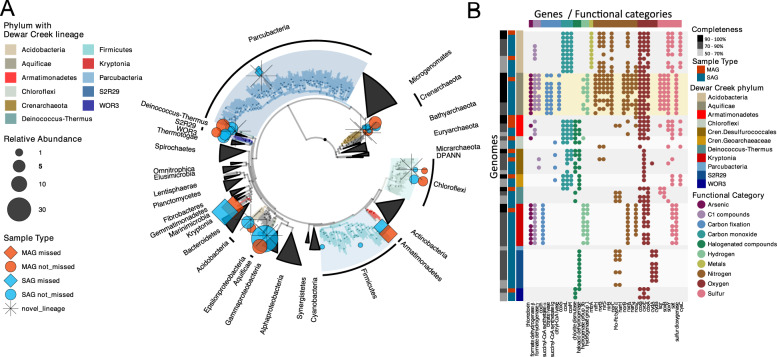
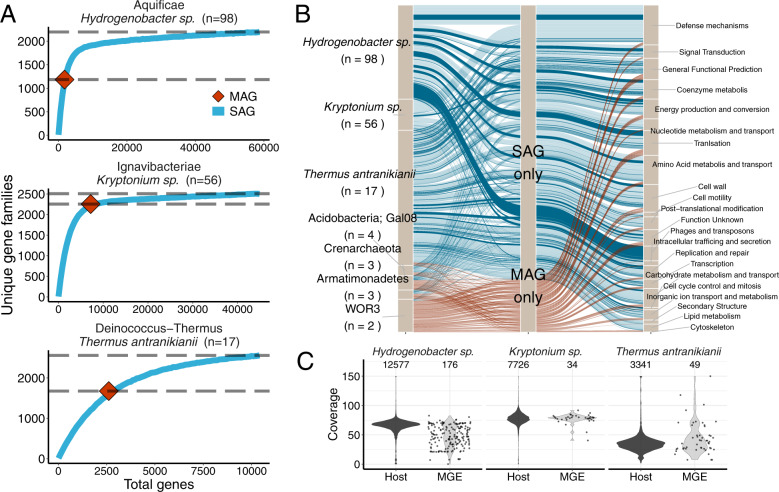
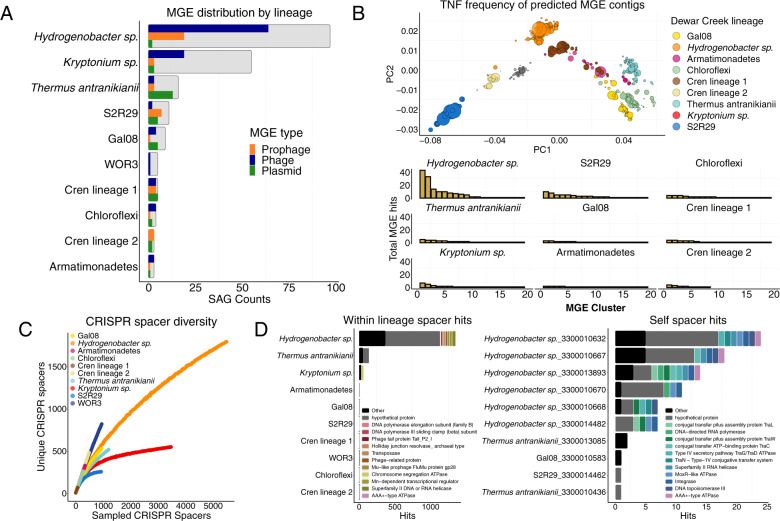
With advances in DNA sequencing and miniaturized molecular biology workflows, rapid and affordable sequencing of single-cell genomes has become a reality. Compared to 16S rRNA gene surveys and shotgun metagenomics, large-scale application of single-cell genomics to whole microbial communities provides an integrated snapshot of community composition and function, directly links mobile elements to their hosts, and enables analysis of population heterogeneity of the dominant community members. To that end, we sequenced nearly 500 single-cell genomes from a low diversity hot spring sediment sample from Dewar Creek, British Columbia, and compared this approach to 16S rRNA gene amplicon and shotgun metagenomics applied to the same sample. We found that the broad taxonomic profiles were similar across the three sequencing approaches, though several lineages were missing from the 16S rRNA gene amplicon dataset, likely the result of primer mismatches. At the functional level, we detected a large array of mobile genetic elements present in the single-cell genomes but absent from the corresponding same species metagenome-assembled genomes. Moreover, we performed a single-cell population genomic analysis of the three most abundant community members, revealing differences in population structure based on mutation and recombination profiles. While the average pairwise nucleotide identities were similar across the dominant species-level lineages, we observed differences in the extent of recombination between these dominant populations. Most intriguingly, the creek's Hydrogenobacter sp. population appeared to be so recombinogenic that it more closely resembled a sexual species than a clonally evolving microbe. Together, this work demonstrates that a randomized single-cell approach can be useful for the exploration of previously uncultivated microbes from community composition to population structure.
随着 DNA 测序和小型化分子生物学工作流程的进步,单细胞基因组的快速和经济测序成为现实。与 16S rRNA 基因调查和鸟枪法宏基因组学相比,单细胞基因组学在整个微生物群落中的大规模应用提供了群落组成和功能的综合快照,直接将移动元件与其宿主联系起来,并能够分析主要群落成员的种群异质性。为此,我们对来自不列颠哥伦比亚省 Dewar Creek 的低多样性温泉沉积物样本中的近 500 个单细胞基因组进行了测序,并将这种方法与应用于相同样本的 16S rRNA 基因扩增子和鸟枪法宏基因组学进行了比较。我们发现,三种测序方法的广泛分类群谱相似,尽管 16S rRNA 基因扩增子数据集缺少几个谱系,可能是引物不匹配的结果。在功能水平上,我们检测到大量存在于单细胞基因组中的移动遗传元件,但在相应的同种宏基因组组装基因组中不存在。此外,我们对三个最丰富的群落成员进行了单细胞群体基因组分析,根据突变和重组谱揭示了种群结构的差异。虽然主导种级谱系的平均成对核苷酸同一性相似,但我们观察到这些优势种群之间重组的程度存在差异。最有趣的是,溪流中的 Hydrogenobacter sp. 种群似乎具有很强的重组性,与无性进化的微生物相比,它更类似于有性物种。总之,这项工作表明,随机单细胞方法可用于探索以前未培养的微生物,从群落组成到种群结构。