Department of Pharmacology, Penn State College of Medicine, Hershey, Pennsylvania 17033, United States.
Department of Biochemistry & Molecular Biology, Penn State College of Medicine, Hershey, Pennsylvania 17033, United States.
J Chem Inf Model. 2022 Feb 14;62(3):463-471. doi: 10.1021/acs.jcim.1c01531. Epub 2022 Feb 1.
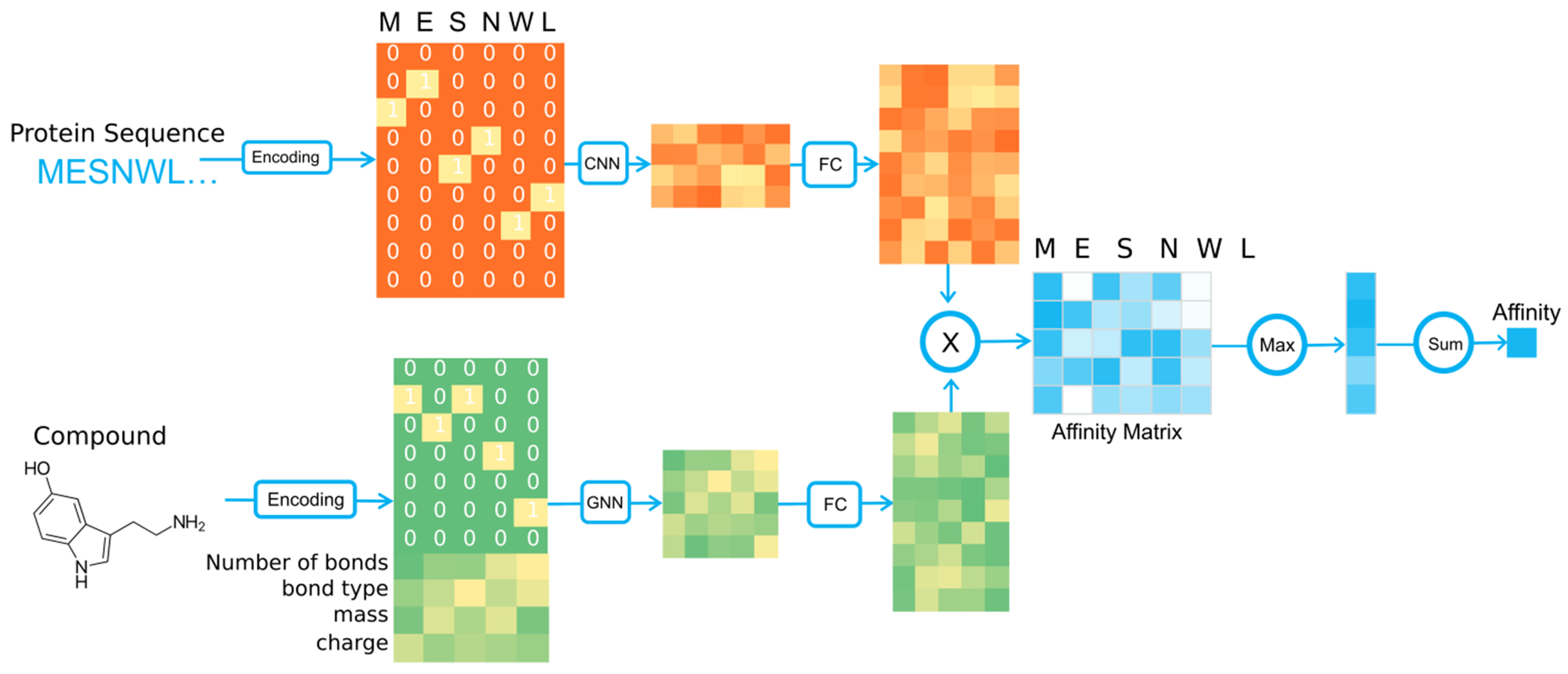
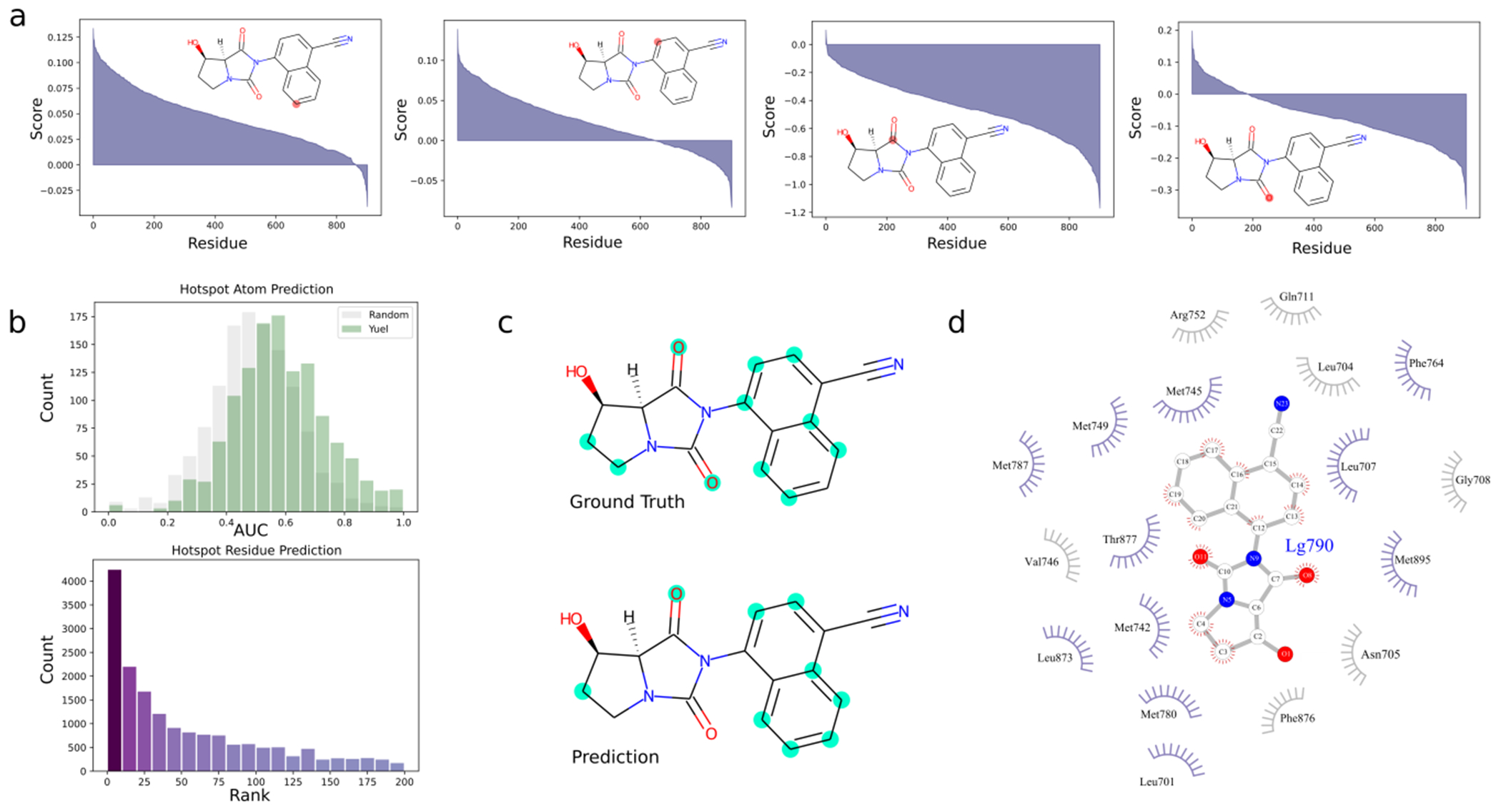
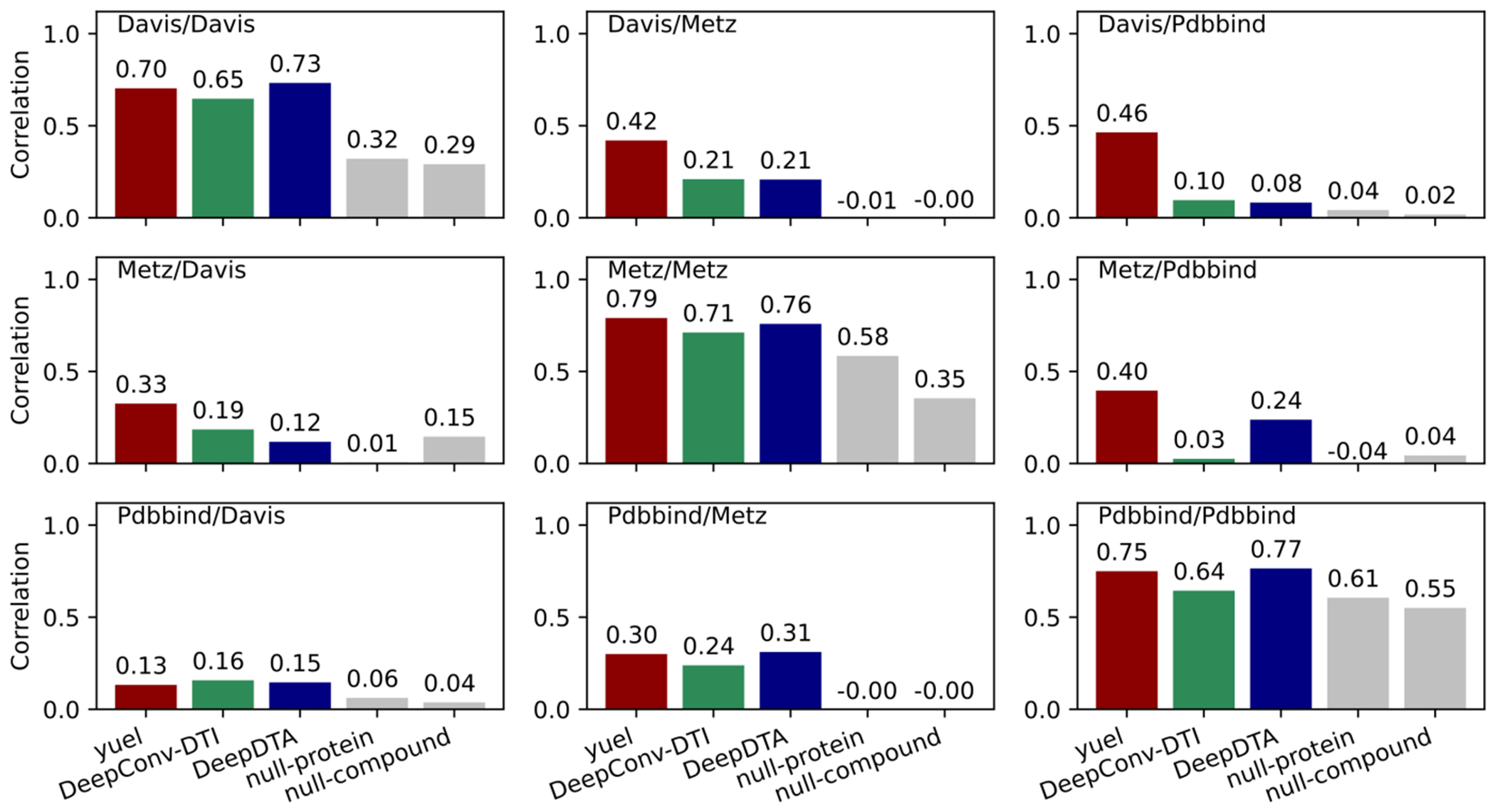
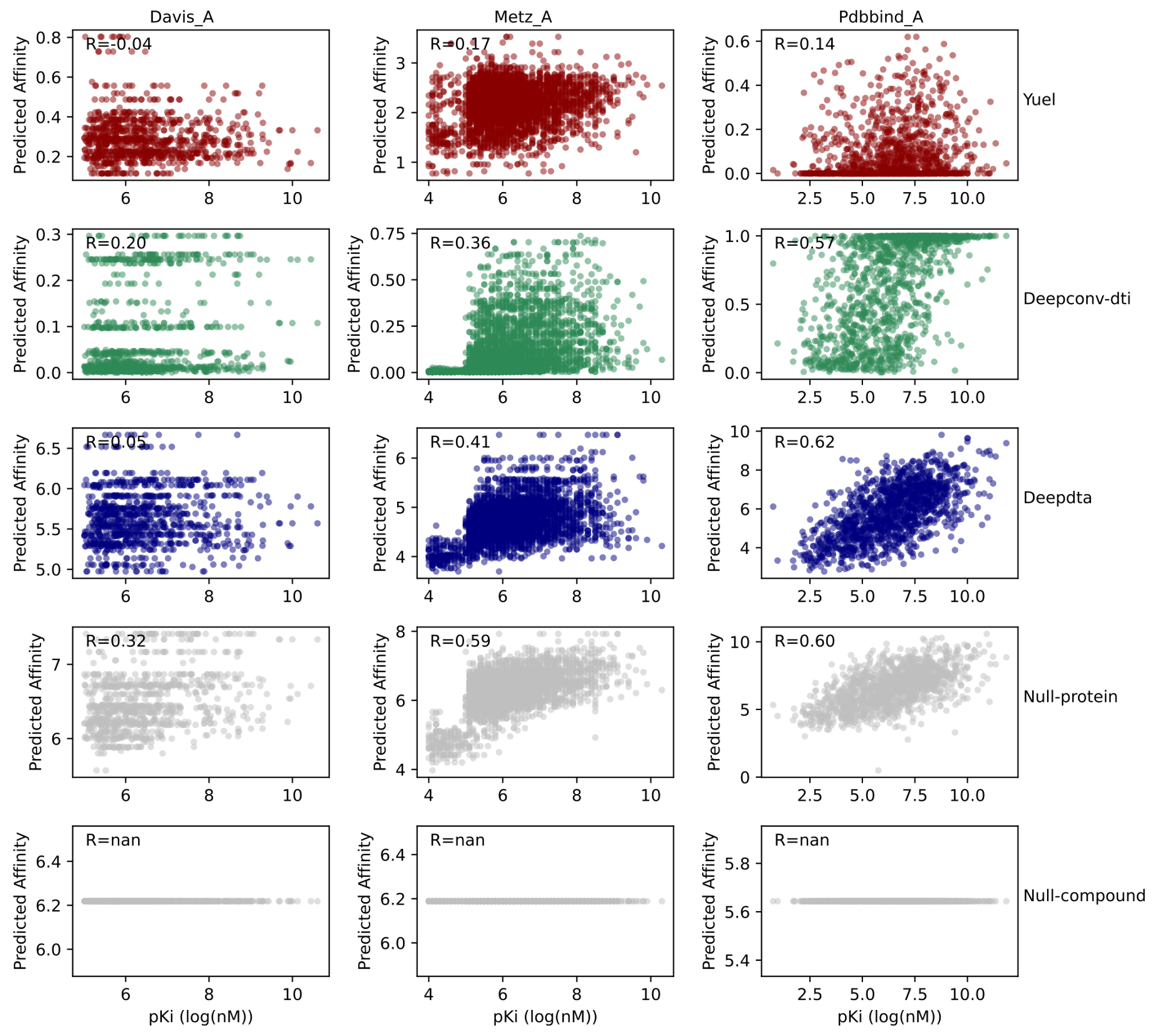
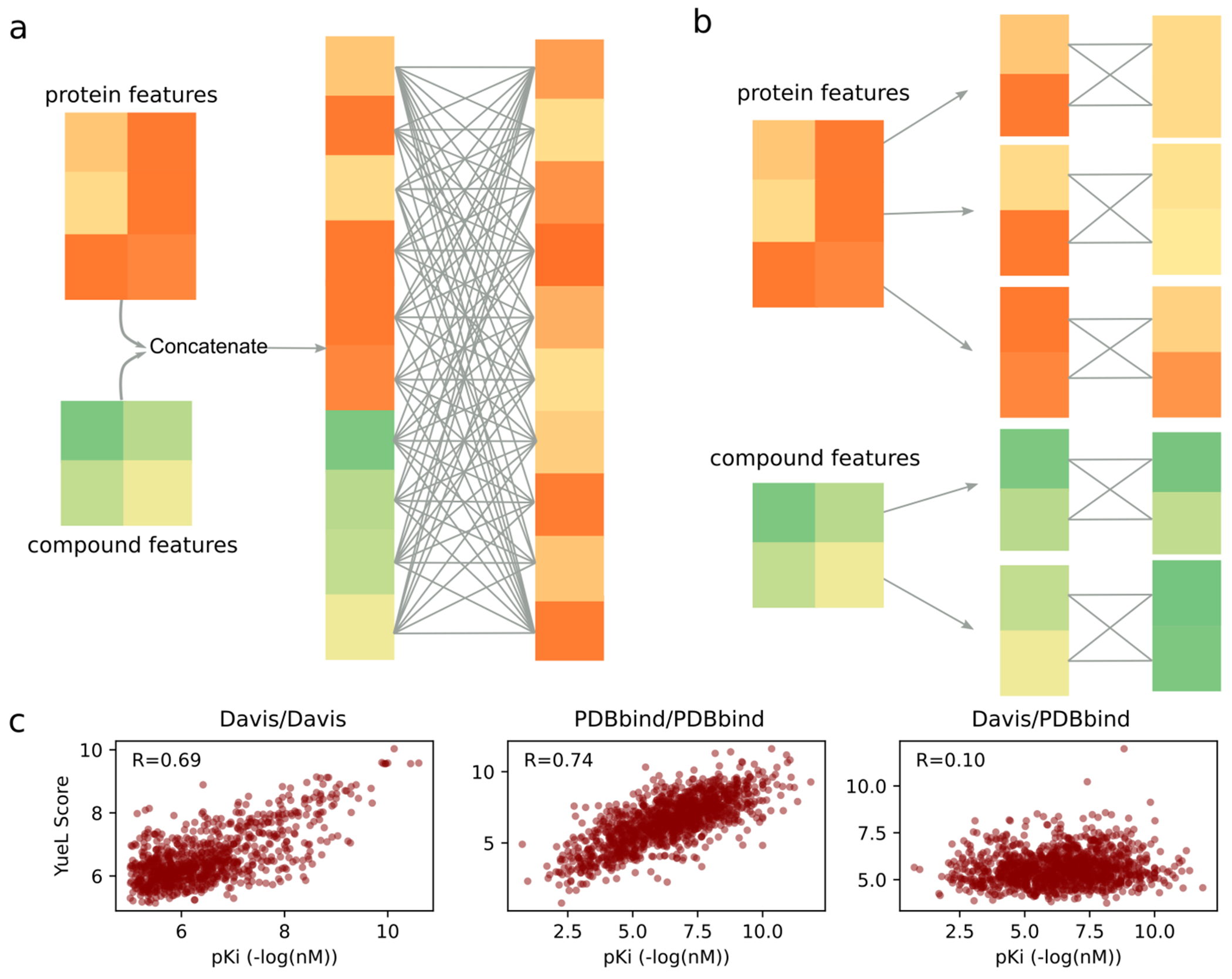
Predicting binding affinities between small molecules and the protein target is at the core of computational drug screening and drug target identification. Deep learning-based approaches have recently been adapted to predict binding affinities and they claim to achieve high prediction accuracy in their tests; we show that these approaches do not generalize, that is, they fail to predict interactions between unknown proteins and unknown small molecules. To address these shortcomings, we develop a new compound-protein interaction predictor, Yuel, which predicts compound-protein interactions with a higher generalizability than the existing methods. Upon comprehensive tests on various data sets, we find that out of all the deep-learning approaches surveyed, Yuel manifests the best ability to predict interactions between unknown compounds and unknown proteins.
预测小分子和蛋白质靶标之间的结合亲和力是计算药物筛选和药物靶标识别的核心。基于深度学习的方法最近被用于预测结合亲和力,并且声称在测试中可以达到很高的预测精度;我们表明这些方法没有泛化能力,也就是说,它们无法预测未知蛋白质和未知小分子之间的相互作用。为了解决这些缺点,我们开发了一种新的化合物-蛋白质相互作用预测器 Yuel,它比现有方法具有更高的通用性,可以预测化合物-蛋白质的相互作用。通过对各种数据集的综合测试,我们发现在所调查的所有深度学习方法中,Yuel 表现出了预测未知化合物和未知蛋白质之间相互作用的最佳能力。