Ye Xia, Gao Qian, Wu Jie, Zhou Lin, Tao Min
Department of Oncology, The First Affiliated Hospital of Soochow University, Suzhou, China.
Transl Cancer Res. 2020 Jul;9(7):4330-4340. doi: 10.21037/tcr-19-2596.
Lung cancer is the most malignant cancer featured with undesirable prognosis. It is urgent to identify novel biomarkers to improve both diagnosis and prognosis. The purpose of the study was to identify significant genes involved in lung cancer through bioinformatic methods and reveal potential underlying mechanisms.
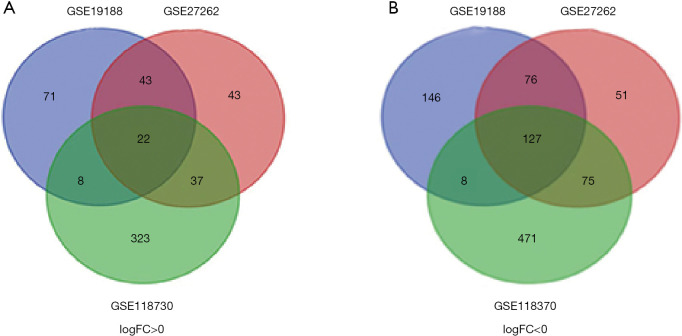
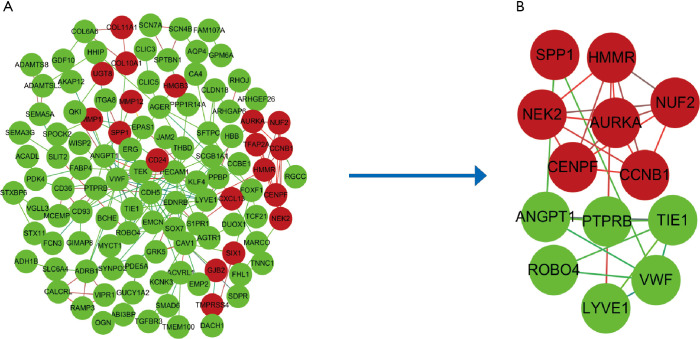
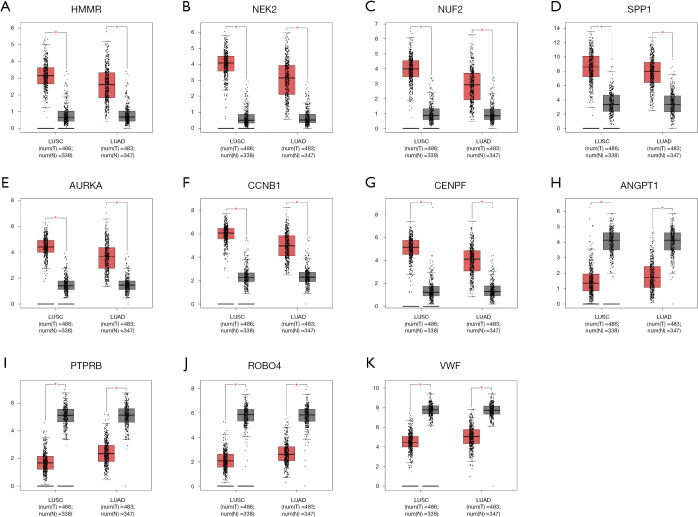
Three datasets GSE19188, GSE27262, GSE118375, containing 122 lung cancer and 96 normal tissues, were available from GEO database. GEO2R and Venn diagram online software were applied to pick out differentially expressed genes (DEGs). Next, we used the Database for Annotation, Visualization and Integrated Discovery (DAVID) to analyze Kyoto Encyclopedia of Gene and Genome (KEGG) pathway and gene ontology (GO) enrichment, followed by protein-protein interaction (PPI) of these DEGs visualized by cytoscape. The MCODE plug-in was performed to construct a module complex of DEGs. In addition, Kaplan-Meier analysis was implemented for analysis of overall survival. To further validate the expression of these genes, Gene Expression Profiling Interactive Analysis (GEPIA) was used.
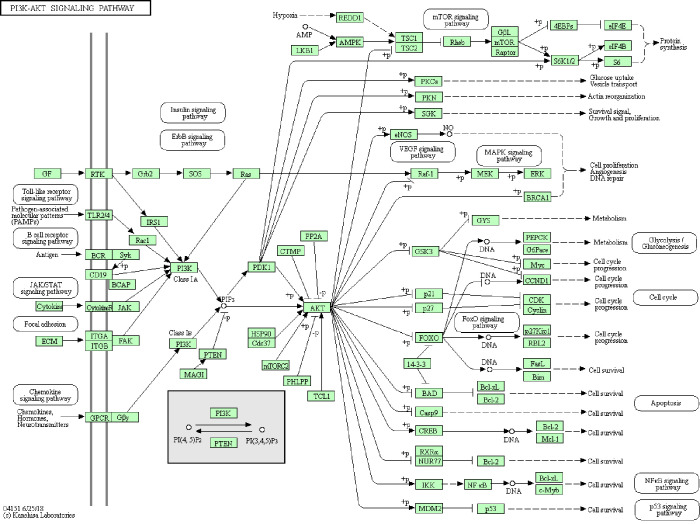
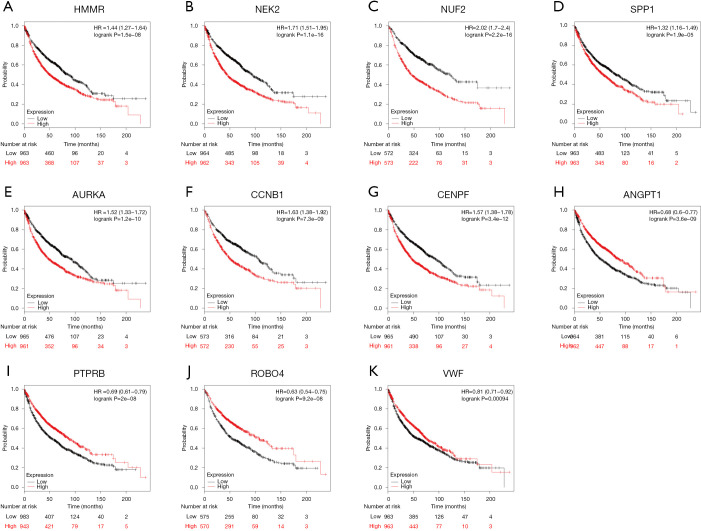
A total of 149 DEGs were identified, including 127 downregulated genes and 22 upregulated genes. KEGG analysis revealed that the DEGs were mainly enriched in ECM-receptor interaction, Vascular smooth muscle contraction, and PPAR signaling pathway. GO analysis of DEGs showed that significant functional enrichment of angiogenesis, cell adhesion, and vasculogenesis. 13 genes were selected as hub genes based on MCODE, and 11 of 13 genes had a significance. The results of GEPIA were consistent with survival analysis. Furthermore, reanalysis of these genes found they were significantly enriched in ECM-receptor interaction and PI3K-Akt signaling pathway.
We have identified several key genes, which could be potential diagnostic and prognostic biomarker as well as therapy targets.
肺癌是最具侵袭性的癌症,预后不良。识别新的生物标志物以改善诊断和预后迫在眉睫。本研究旨在通过生物信息学方法识别参与肺癌的重要基因,并揭示潜在的机制。
从GEO数据库获取三个数据集GSE19188、GSE27262、GSE118375,包含122例肺癌组织和96例正常组织。使用GEO2R和Venn图在线软件筛选差异表达基因(DEG)。接下来,我们使用注释、可视化和综合发现数据库(DAVID)分析京都基因与基因组百科全书(KEGG)通路和基因本体(GO)富集,随后通过Cytoscape对这些DEG进行蛋白质-蛋白质相互作用(PPI)可视化。使用MCODE插件构建DEG的模块复合体。此外,采用Kaplan-Meier分析评估总生存期。为进一步验证这些基因的表达,使用基因表达谱交互式分析(GEPIA)。
共鉴定出149个DEG,包括127个下调基因和22个上调基因。KEGG分析显示,DEG主要富集于细胞外基质-受体相互作用、血管平滑肌收缩和PPAR信号通路。DEG的GO分析表明,血管生成、细胞黏附及血管发生存在显著功能富集。基于MCODE选择13个基因作为枢纽基因,其中11个具有显著性。GEPIA结果与生存分析一致。此外,对这些基因的重新分析发现它们在细胞外基质-受体相互作用和PI3K-Akt信号通路中显著富集。
我们已鉴定出几个关键基因,它们可能是潜在的诊断和预后生物标志物以及治疗靶点。