Department of Computational Medicine and Bioinformatics, University of Michigan, Ann Arbor, MI, USA.
Department of Computer Science and Engineering, University of Michigan, Ann Arbor, MI, USA.
Nat Commun. 2022 Feb 9;13(1):780. doi: 10.1038/s41467-022-28431-4.
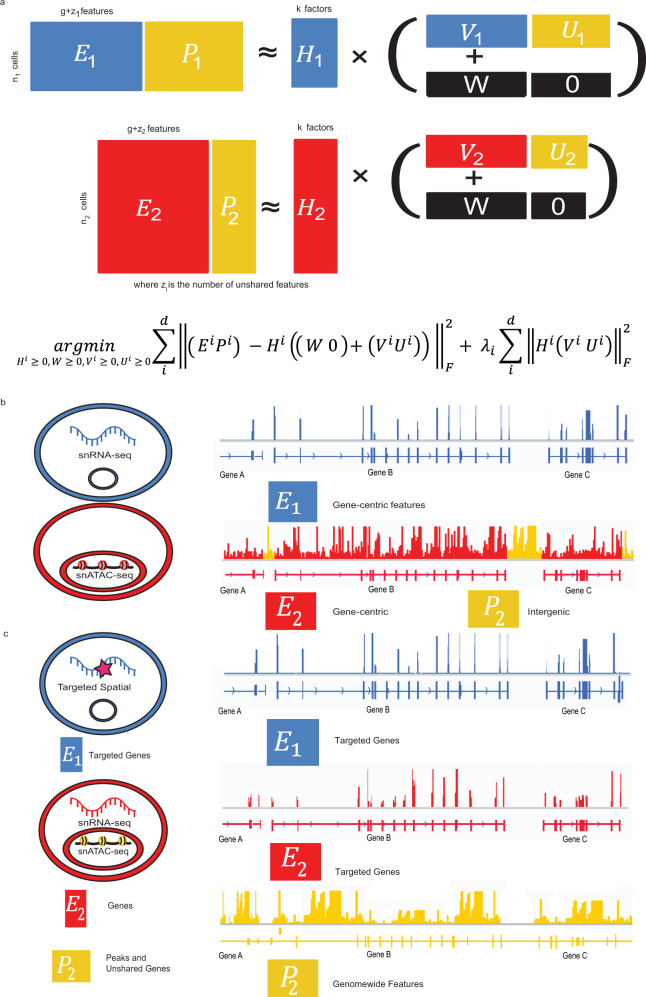
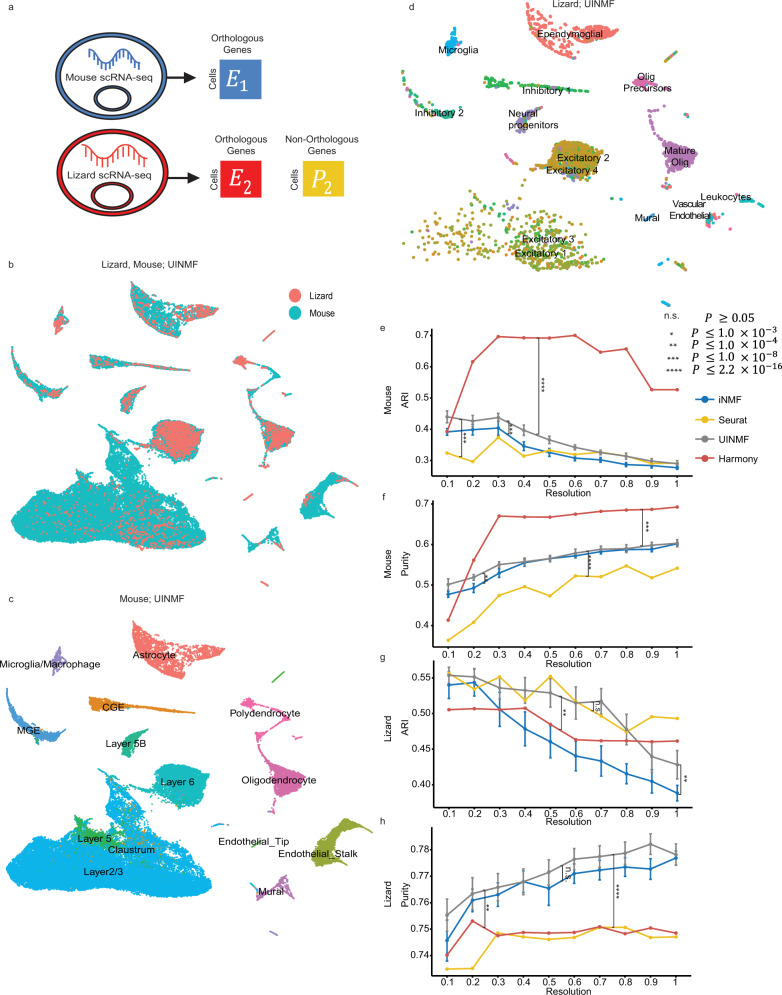
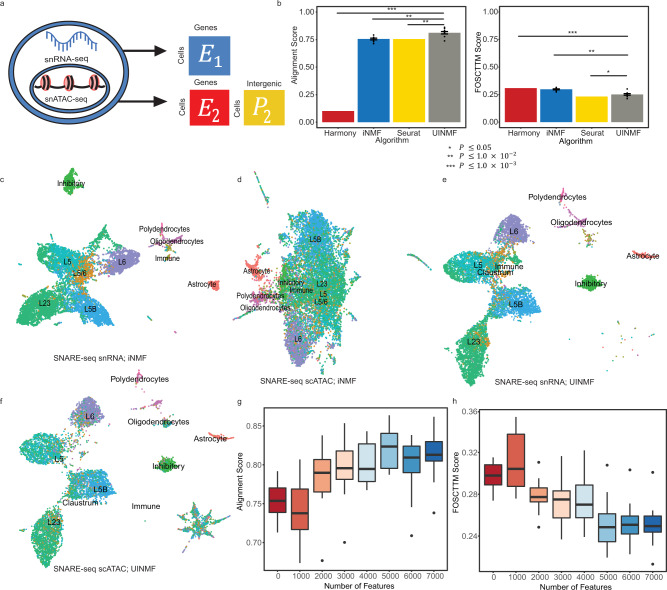
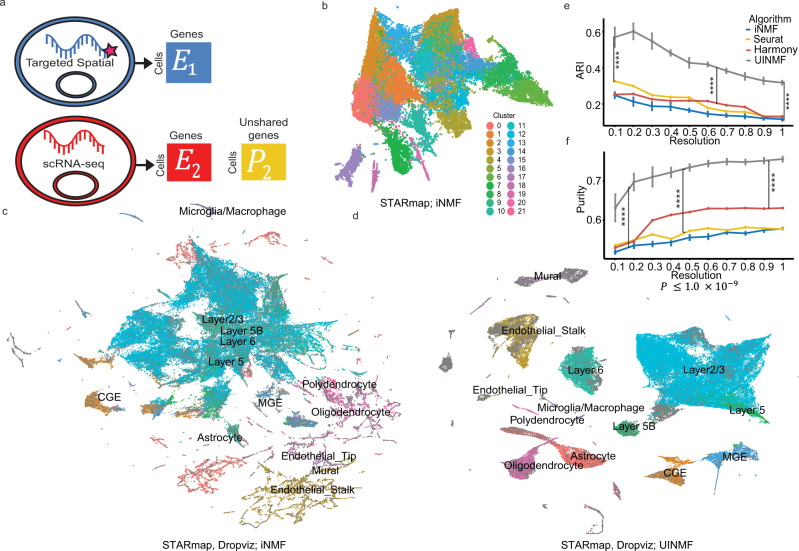
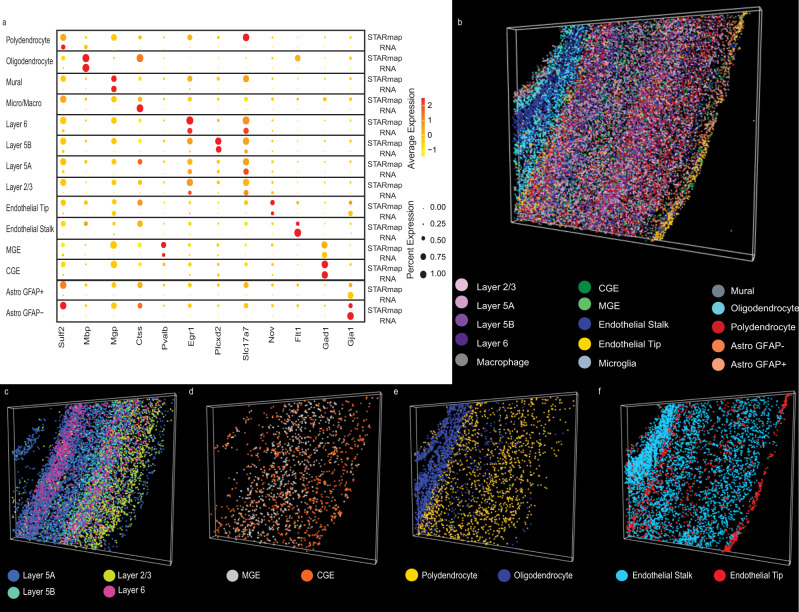
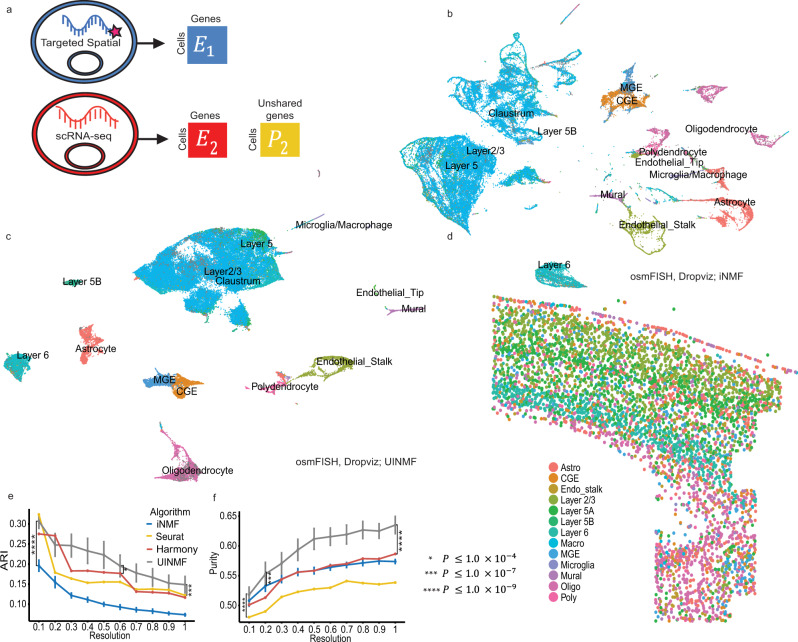
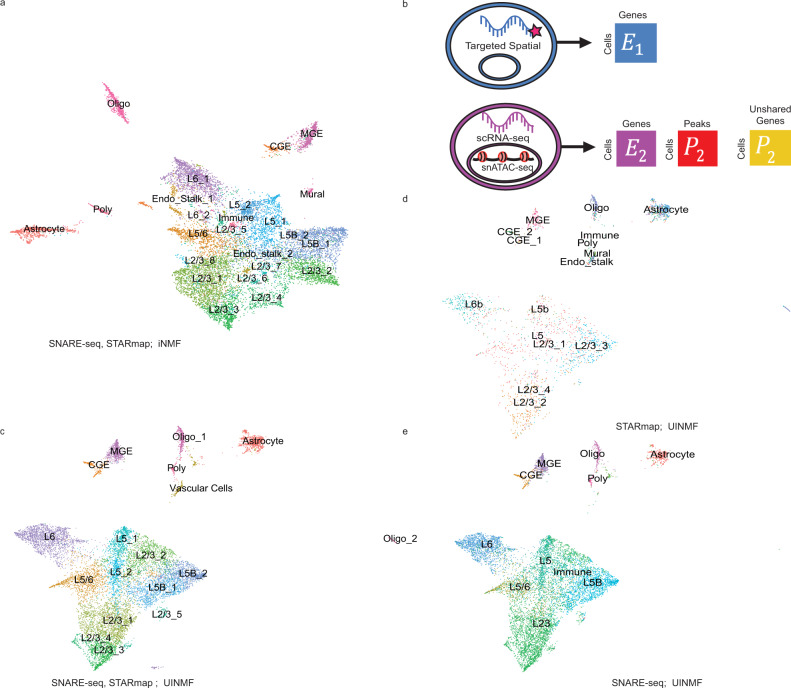
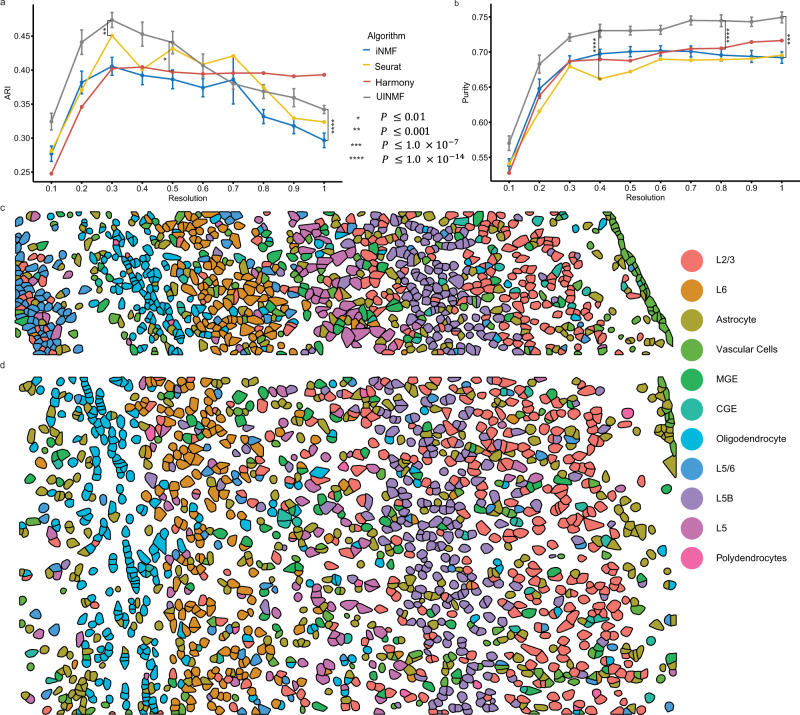
Single-cell genomic technologies provide an unprecedented opportunity to define molecular cell types in a data-driven fashion, but present unique data integration challenges. Many analyses require "mosaic integration", including both features shared across datasets and features exclusive to a single experiment. Previous computational integration approaches require that the input matrices share the same number of either genes or cells, and thus can use only shared features. To address this limitation, we derive a nonnegative matrix factorization algorithm for integrating single-cell datasets containing both shared and unshared features. The key advance is incorporating an additional metagene matrix that allows unshared features to inform the factorization. We demonstrate that incorporating unshared features significantly improves integration of single-cell RNA-seq, spatial transcriptomic, SNARE-seq, and cross-species datasets. We have incorporated the UINMF algorithm into the open-source LIGER R package ( https://github.com/welch-lab/liger ).
单细胞基因组技术提供了一个前所未有的机会,可以以数据驱动的方式定义分子细胞类型,但也带来了独特的数据集成挑战。许多分析需要“镶嵌式集成”,包括跨数据集共享的特征和单个实验特有的特征。以前的计算集成方法要求输入矩阵共享相同数量的基因或细胞,因此只能使用共享特征。为了解决这个限制,我们推导出了一种非负矩阵分解算法,用于集成包含共享和非共享特征的单细胞数据集。关键的进展是引入了一个额外的元基因矩阵,允许非共享特征为分解提供信息。我们证明了纳入非共享特征可以显著改善单细胞 RNA-seq、空间转录组学、SNARE-seq 和跨物种数据集的集成。我们已经将 UINMF 算法纳入了开源 LIGER R 包(https://github.com/welch-lab/liger)。