Graduate Program in Biotechnology and Biodiversity-Network BIONORTE, Federal University of Amapá, Macapá 68903-419, AP, Brazil.
Laboratory of Modeling and Computational Chemistry, Department of Biological and Health Sciences, Federal University of Amapá, Macapá 68902-280, AP, Brazil.
Int J Mol Sci. 2022 Feb 4;23(3):1781. doi: 10.3390/ijms23031781.
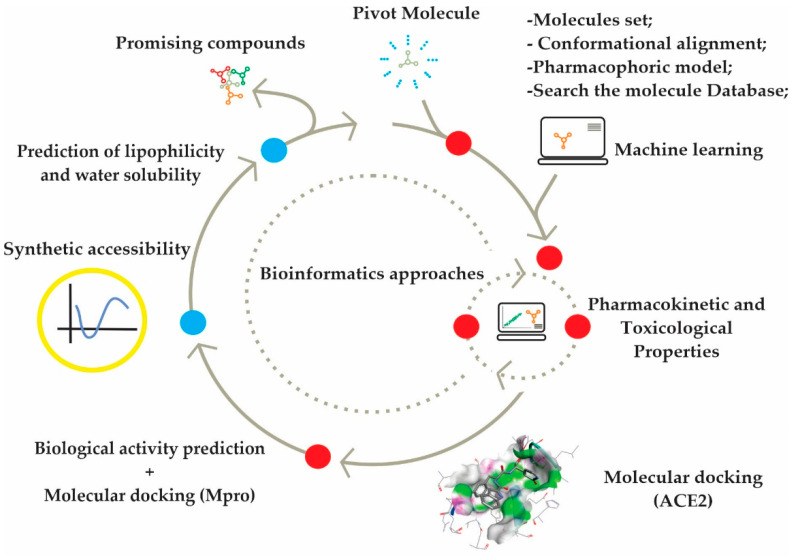
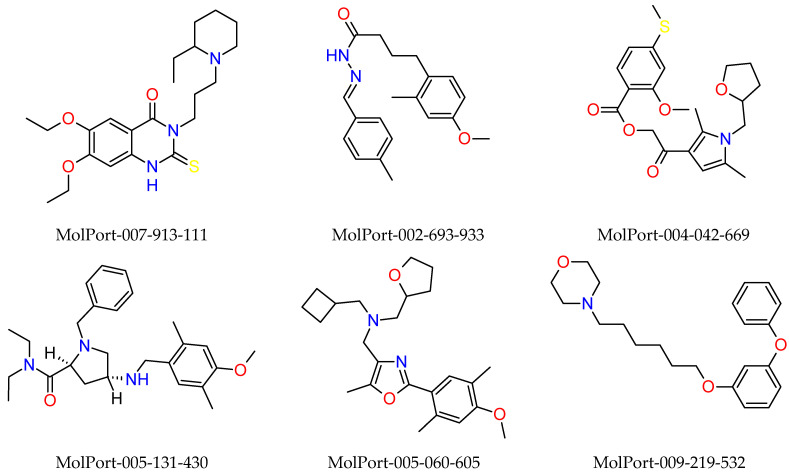
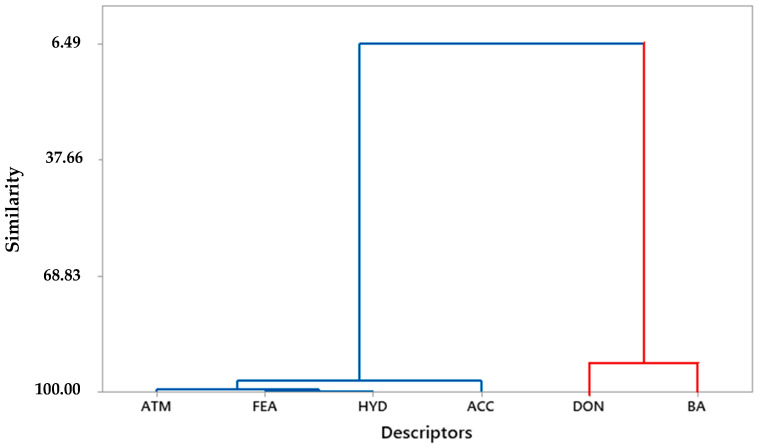
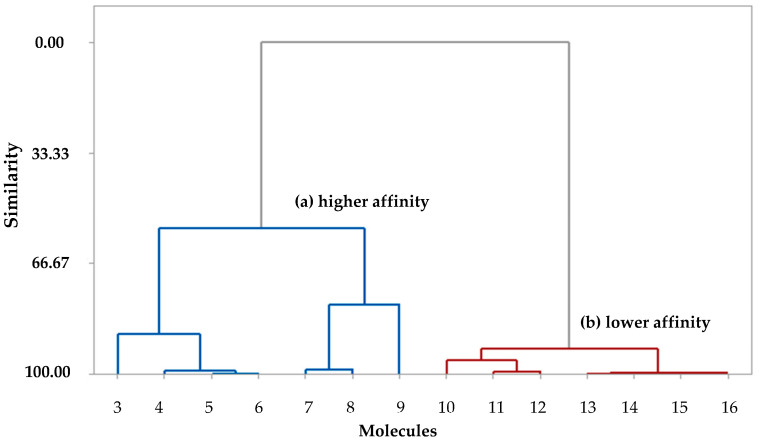
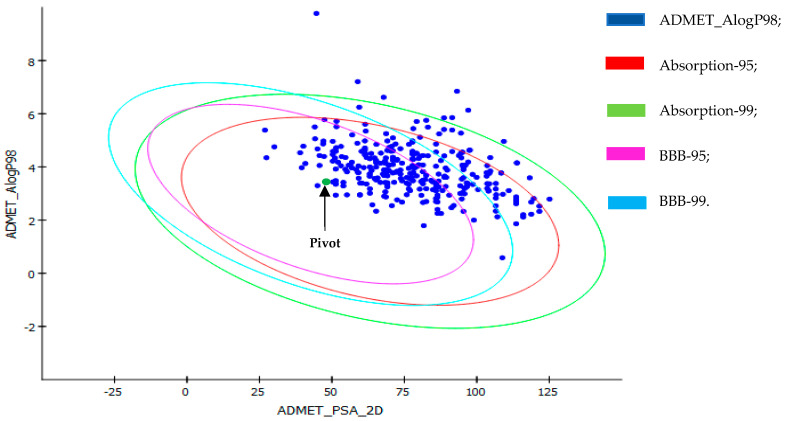
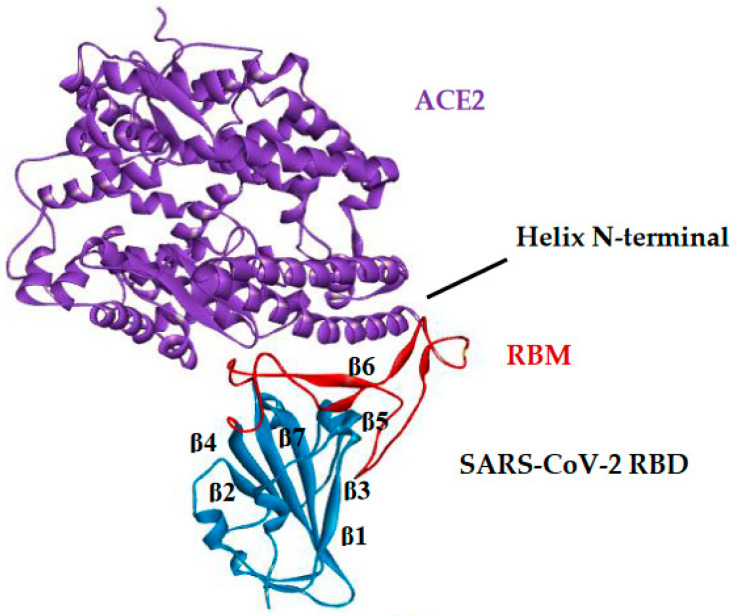
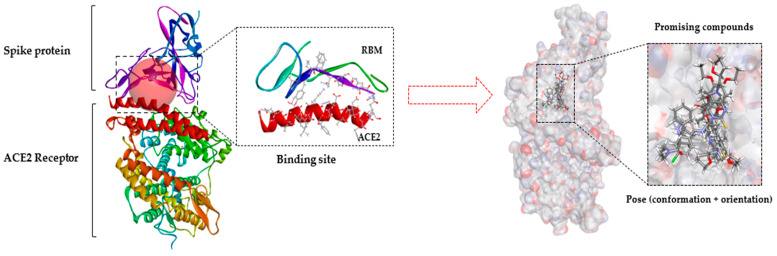
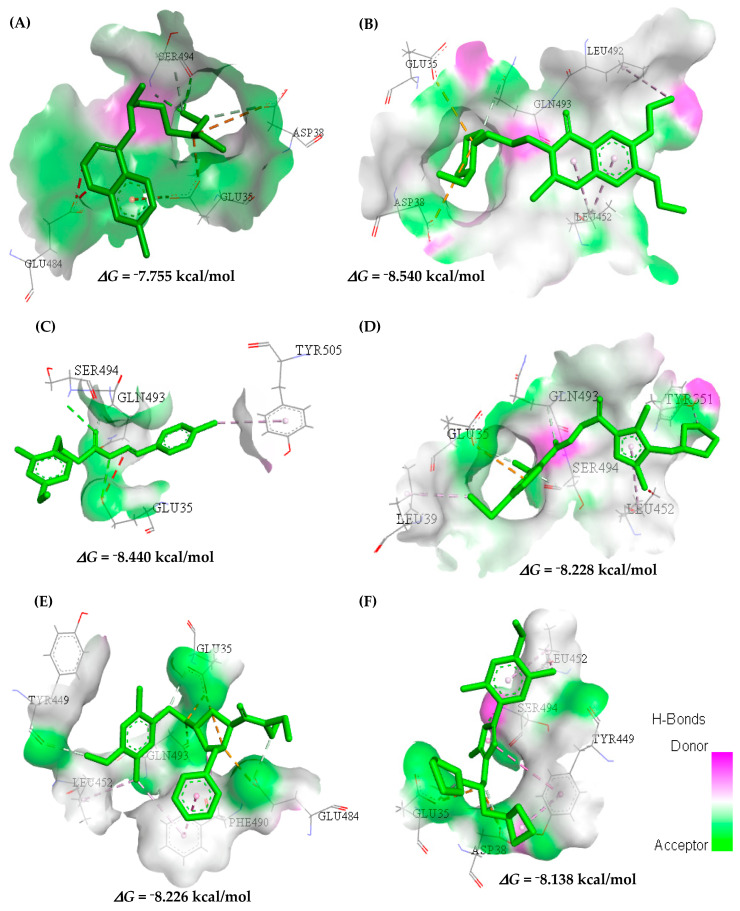
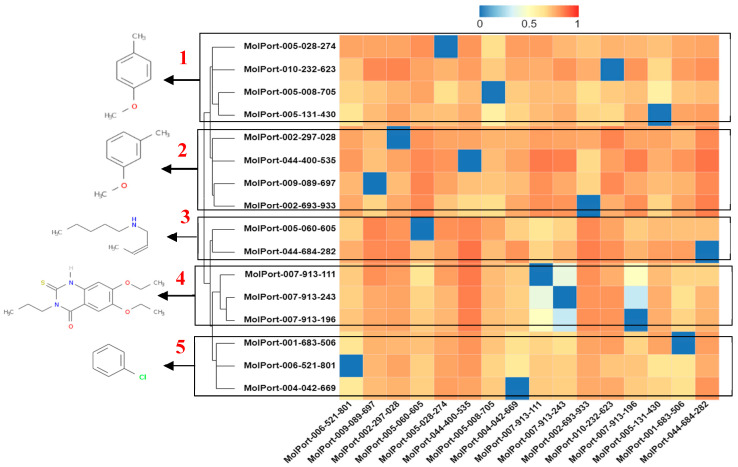
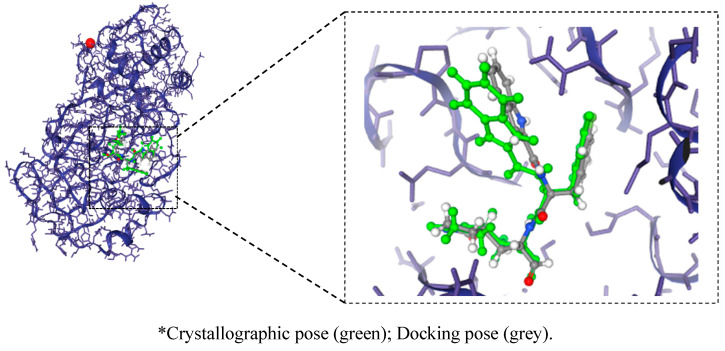
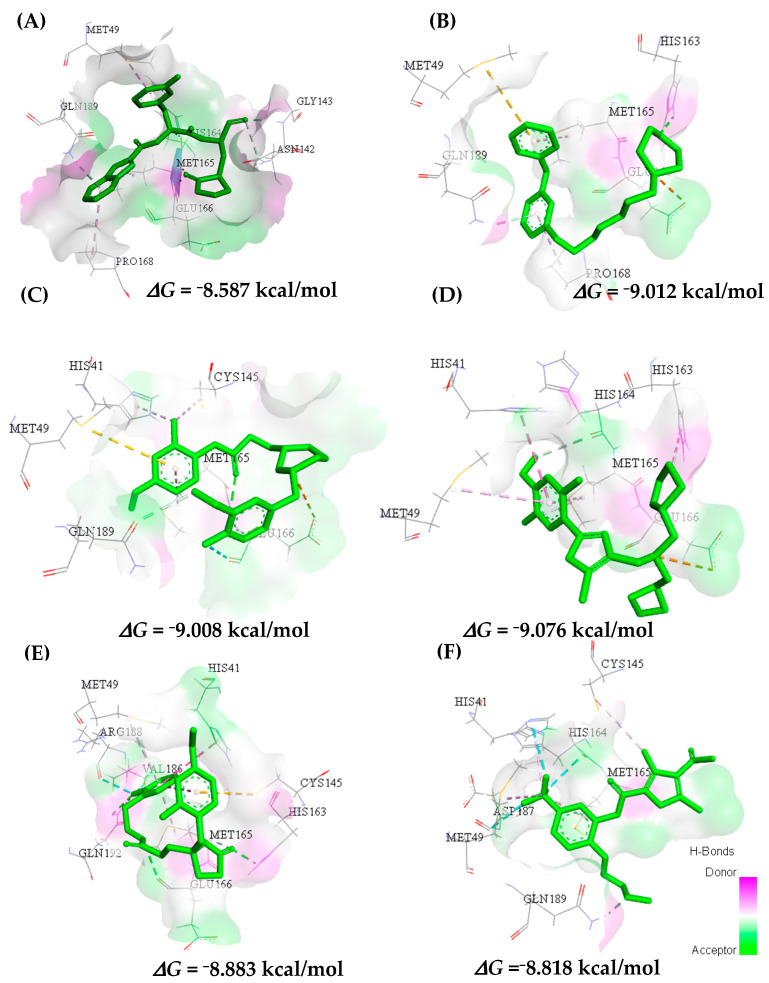
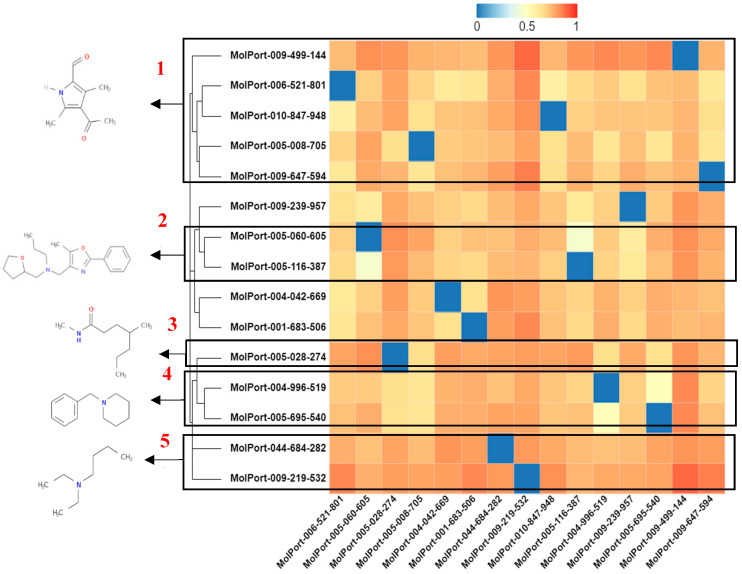
This study aimed to identify potential inhibitors and investigate the mechanism of action on SARS-CoV-2 ACE2 receptors using a molecular modeling study and theoretical determination of biological activity. Hydroxychloroquine was used as a pivot structure and antimalarial analogues of 1,2,4,5 tetraoxanes were used for the construction and evaluation of pharmacophoric models. The pharmacophore-based virtual screening was performed on the Molport database (~7.9 million compounds) and obtained 313 structures. Additionally, a pharmacokinetic study was developed, obtaining 174 structures with 99% confidence for human intestinal absorption and penetration into the blood-brain barrier (BBB); posteriorly, a study of toxicological properties was realized. Toxicological predictions showed that the selected molecules do not present a risk of hepatotoxicity, carcinogenicity, mutagenicity, and skin irritation. Only 54 structures were selected for molecular docking studies, and five structures showed binding affinity (ΔG) values satisfactory for ACE2 receptors (PDB 6M0J), in which the molecule MolPort-007-913-111 had the best ΔG value of -8.540 Kcal/mol, followed by MolPort-002-693-933 with ΔG = -8.440 Kcal/mol. Theoretical determination of biological activity was realized for 54 structures, and five molecules showed potential protease inhibitors. Additionally, we investigated the Mpro receptor (6M0K) for the five structures via molecular docking, and we confirmed the possible interaction with the target. In parallel, we selected the TopsHits 9 with antiviral potential that evaluated synthetic accessibility for future synthesis studies and in vivo and in vitro tests.
本研究旨在使用分子建模研究和理论生物活性测定来鉴定潜在的抑制剂,并研究其对 SARS-CoV-2 ACE2 受体的作用机制。以羟氯喹为枢纽结构,以 1,2,4,5-四氧杂环戊烷的抗疟类似物为构建和评价药效团模型的基础。基于药效团的虚拟筛选在 Molport 数据库(~790 万种化合物)上进行,得到 313 种结构。此外,还进行了药代动力学研究,获得了 174 种具有 99%置信度的人类肠道吸收和穿透血脑屏障(BBB)的结构;随后,进行了毒理学性质研究。毒理学预测表明,所选分子不存在肝毒性、致癌性、致突变性和皮肤刺激性风险。只有 54 种结构被选用于分子对接研究,其中 5 种结构显示出与 ACE2 受体的结合亲和力(ΔG)值令人满意(PDB 6M0J),其中分子 MolPort-007-913-111 的 ΔG 值最佳,为-8.540 Kcal/mol,其次是 MolPort-002-693-933,ΔG=-8.440 Kcal/mol。对 54 种结构进行了理论生物活性测定,其中 5 种分子表现出潜在的蛋白酶抑制剂活性。此外,我们通过分子对接研究了这 5 种结构的 Mpro 受体(6M0K),并证实了与靶标的可能相互作用。同时,我们选择了具有抗病毒潜力的 Topshits9,评估了其合成可及性,以便进行未来的合成研究以及体内和体外测试。