National Clinical Research Center for Infectious Diseases, Shenzhen Third People's Hospital, Southern University of Science and Technology, Shenzhen, China.
Guangdong Key Laboratory of Regional Immunity and Diseases, Shenzhen University School of Medicine, Shenzhen, China.
Microbiol Spectr. 2022 Feb 23;10(1):e0190121. doi: 10.1128/spectrum.01901-21.
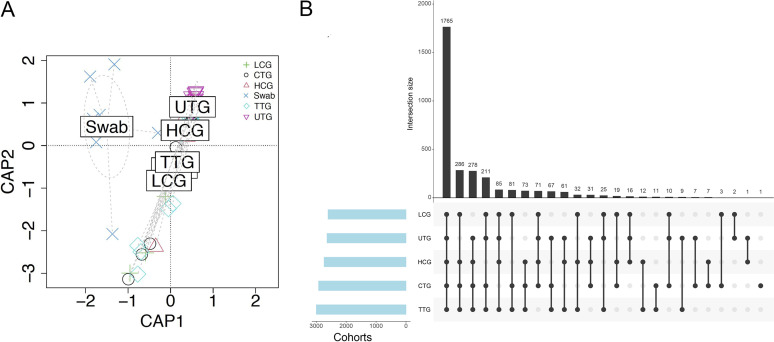
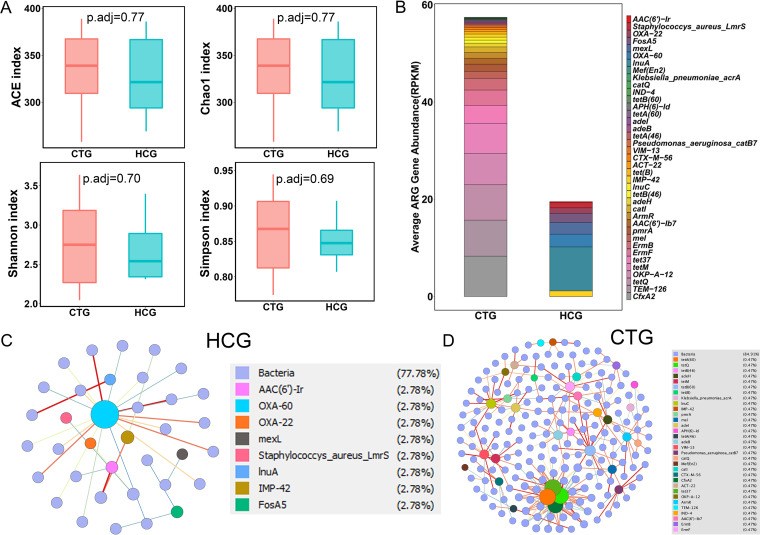
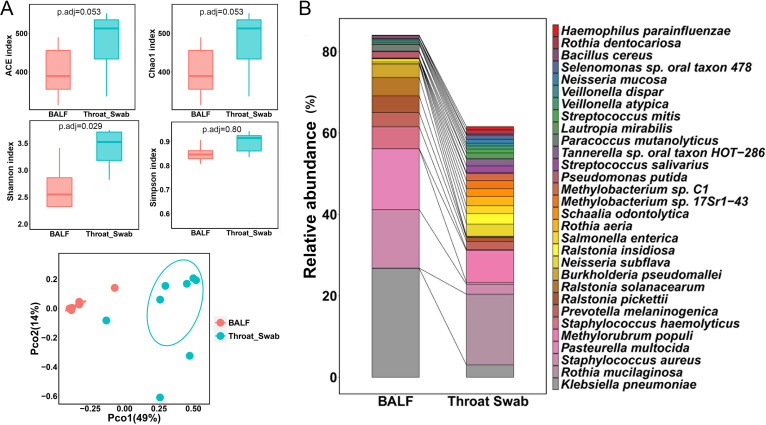
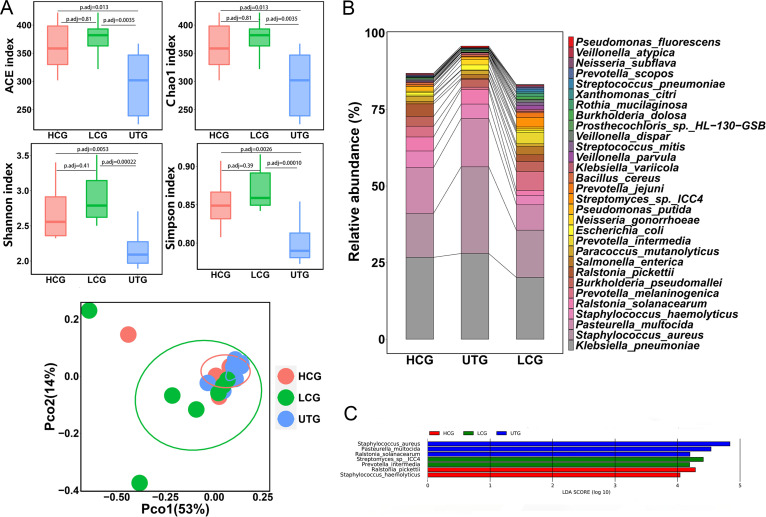
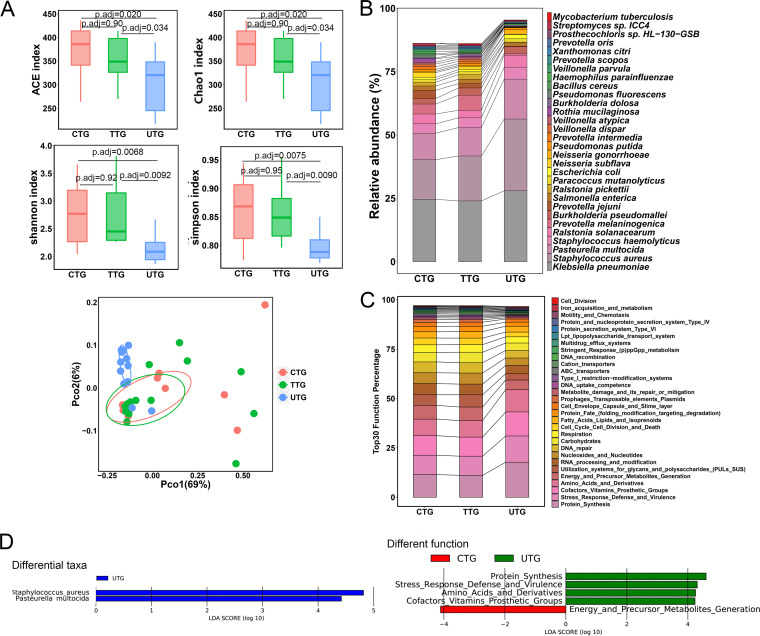
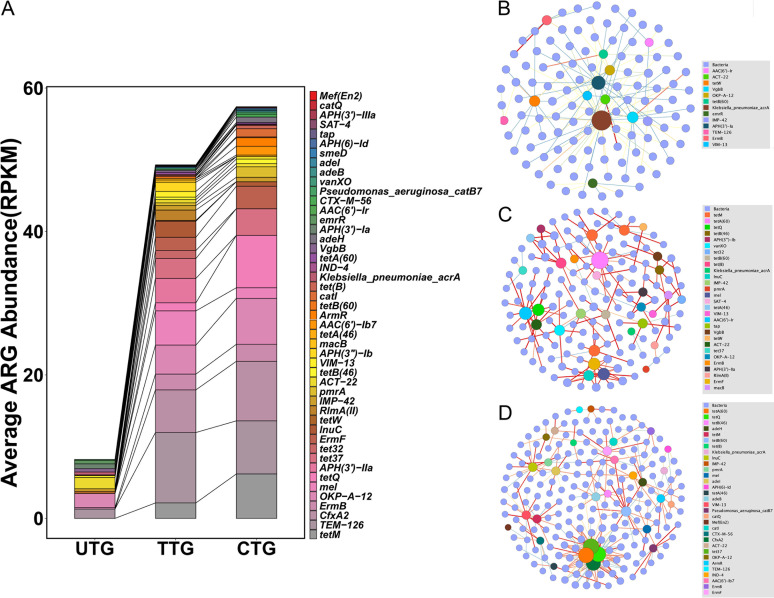
The microbiota plays an important role in human health and disease development. The lung microbiota profile in pulmonary tuberculosis (TB) patients and the effects of anti-TB treatment on the profile need to be determined thoroughly and comprehensively. This study primarily aimed to determine the lung microbiota profile associated with pulmonary TB and characterize the longitudinal changes during anti-TB treatment. A total of 53 participants, comprising 8 healthy individuals, 12 untreated pulmonary TB patients, 15 treated pulmonary TB patients, 11 cured pulmonary TB patients, and 7 lung cancer patients, were recruited in the present study. Bronchioalveolar lavage fluid (BALF) samples were collected from the above participants, and throat swabs were taken from healthy individuals. Microbiomes in the samples were examined using metagenomic next-generation sequencing (mNGS). Differences in microbiota profiles were determined through a comparison of the indicated groups. Our findings indicated that the BALF samples displayed decreased richness and diversity of the microbiota compared to those of the throat swab samples, and these two kinds of samples exhibited obvious separation on principal-coordinate analysis (PCoA) plots. Untreated pulmonary TB patients displayed a unique lung microbiota signature distinct from that of healthy individuals and lung cancer patients. Our data first demonstrated that anti-TB treatment with first-line drugs increases alpha diversity and significantly affects the beta diversity of the lung microbiota, while it also induces antibiotic resistance genes (ARGs). Characterization of the lung microbiota could lead to a better understanding of the pathogenesis of pulmonary TB. Here, we applied the metagenomic shotgun sequencing instead of 16S rRNA sequencing method to characterize the lung microbiota using the BALF samples instead of sputum. We found that alterations in the lung microbiota are associated with TB infection and that anti-TB treatment significantly affects the alpha and beta diversity of the lung microbiota in pulmonary TB patients. These findings could help us better understand TB pathogenesis.
微生物组在人类健康和疾病发展中发挥着重要作用。需要彻底和全面地确定肺结核(TB)患者肺部微生物组的特征以及抗 TB 治疗对其特征的影响。本研究主要旨在确定与肺结核相关的肺部微生物组特征,并描述抗 TB 治疗过程中的纵向变化。本研究共招募了 53 名参与者,包括 8 名健康个体、12 名未经治疗的肺结核患者、15 名接受治疗的肺结核患者、11 名已治愈的肺结核患者和 7 名肺癌患者。从上述参与者中采集支气管肺泡灌洗液(BALF)样本,并从健康个体中采集咽拭子样本。使用宏基因组下一代测序(mNGS)检查样本中的微生物组。通过比较指定组确定微生物组谱的差异。我们的研究结果表明,与咽拭子样本相比,BALF 样本中的微生物组丰富度和多样性降低,这两种样本在主坐标分析(PCoA)图谱上明显分离。未经治疗的肺结核患者表现出与健康个体和肺癌患者不同的独特肺部微生物组特征。我们的数据首次表明,一线药物抗 TB 治疗可增加 alpha 多样性,并显著影响肺部微生物组的 beta 多样性,同时还会诱导抗生素耐药基因(ARGs)。对肺部微生物组的特征进行描述可以更好地了解肺结核的发病机制。在这里,我们应用宏基因组鸟枪法测序而不是 16S rRNA 测序方法,使用 BALF 样本而不是痰液来描述肺部微生物组。我们发现,肺部微生物组的变化与 TB 感染有关,抗 TB 治疗显著影响肺结核患者肺部微生物组的 alpha 和 beta 多样性。这些发现可以帮助我们更好地了解结核病的发病机制。