Department of Thoracic Surgery, The Second Affiliated Hospital of Harbin Medical University, Harbin, China.
Burning Rock Biotech, Guangzhou, China.
BMC Med Genomics. 2022 Mar 21;15(1):66. doi: 10.1186/s12920-022-01192-1.
Field cancerization is the process in which a population of normal or pre-malignant cells is affected by oncogenic alterations leading to progressive molecular changes that drive malignant transformation. Aberrant DNA methylation has been implicated in early cancer development in non-small cell lung cancer (NSCLC); however, studies on its role in field cancerization (FC) are limited. This study aims to identify FC-specific methylation patterns that could distinguish between pre-malignant lesions and tumor tissues in NSCLC.
We enrolled 52 patients with resectable NSCLC and collected resected tumor (TUM), tumor-adjacent (ADJ) and tumor-distant normal (DIS) tissue samples, among whom 36 qualified for subsequent analyses. Methylation levels were profiled by bisulfite sequencing using a custom lung-cancer methylation panel.
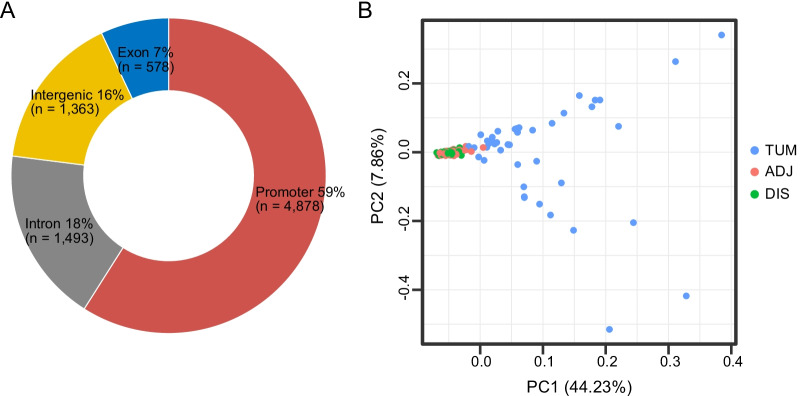
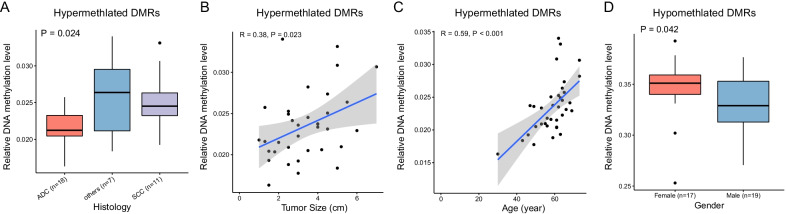
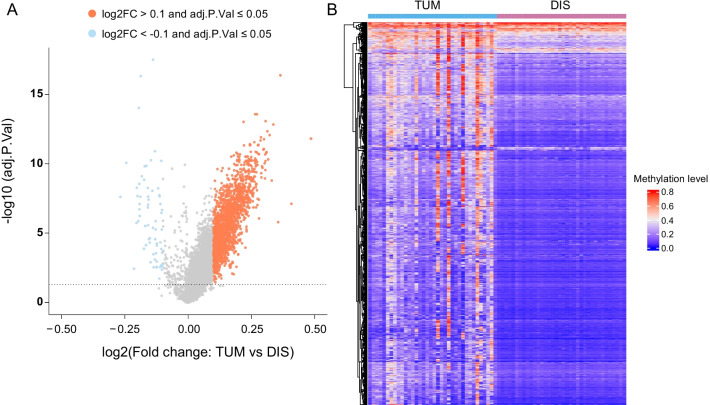
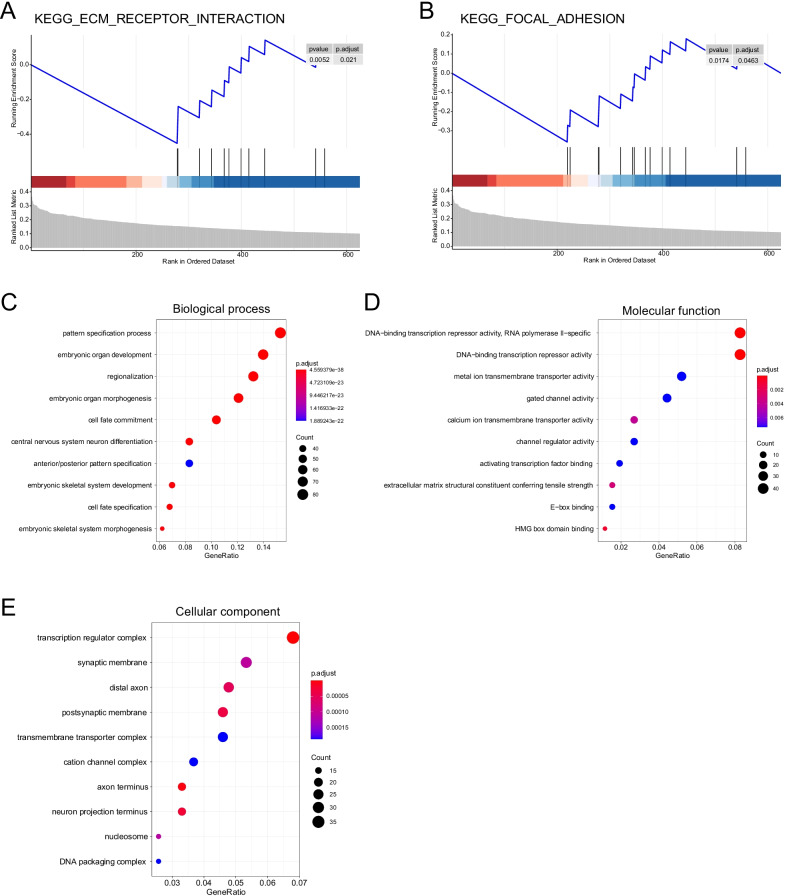
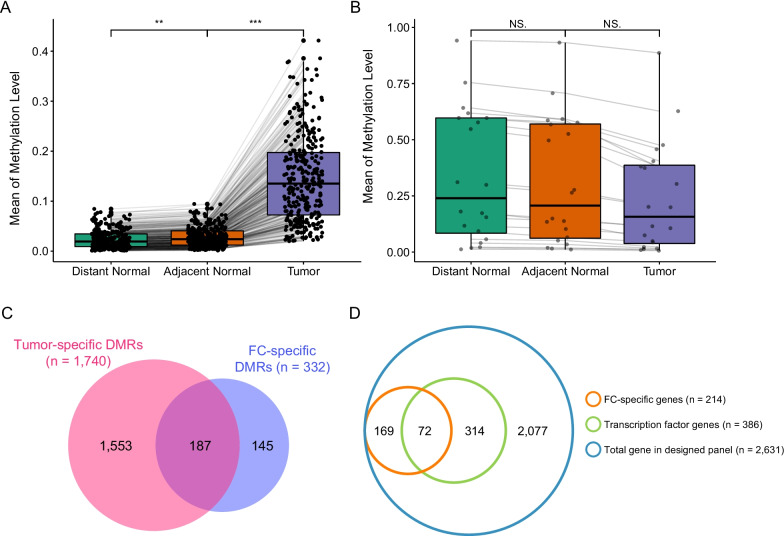
ADJ and DIS samples demonstrated similar methylation profiles, which were distinct from distinct from that of TUM. Comparison of TUM and DIS profiles led to identification of 1740 tumor-specific differential methylated regions (DMRs), including 1675 hypermethylated and 65 hypomethylated (adjusted P < 0.05). Six of the top 10 tumor-specific hypermethylated regions were associated with cancer development. We then compared the TUM, ADJ, and DIS to further identify the progressively aggravating aberrant methylations during cancer initiation and early development. A total of 332 DMRs were identified, including a predominant proportion of 312 regions showing stepwise increase in methylation levels as the sample drew nearer to the tumor (i.e. DIS < ADJ < TUM) and 20 regions showing a stepwise decrease pattern. Gene set enrichment analysis (GSEA) for KEGG and GO terms consistently suggested enrichment of DMRs located in transcription factor genes, suggesting a central role of epigenetic regulation of transcription factors in FC and tumorigenesis.
We revealed distinct methylation patterns between pre-malignant lesions and malignant tumors, suggesting the essential role of DNA methylation as an early step in pre-malignant field defects. Moreover, our study also identified differentially methylated genes, especially transcription factors, that could potentially be used as markers for lung cancer screening and for mechanistic studies of FC and early cancer development.
田间癌变是指一群正常或癌前细胞受到致癌改变的影响,导致进行性的分子变化,从而驱动恶性转化的过程。异常的 DNA 甲基化已被发现在非小细胞肺癌 (NSCLC) 的早期癌症发展中起作用; 然而,关于其在田间癌变 (FC) 中的作用的研究有限。本研究旨在确定能够区分 NSCLC 癌前病变和肿瘤组织的 FC 特异性甲基化模式。
我们招募了 52 名可切除 NSCLC 患者,并收集了切除的肿瘤 (TUM)、肿瘤相邻 (ADJ) 和肿瘤远处正常 (DIS) 组织样本,其中 36 名符合后续分析条件。使用定制的肺癌甲基化面板通过亚硫酸氢盐测序对甲基化水平进行了分析。
ADJ 和 DIS 样本表现出相似的甲基化谱,与 TUM 明显不同。比较 TUM 和 DIS 图谱导致鉴定出 1740 个肿瘤特异性差异甲基化区域 (DMRs),包括 1675 个高甲基化和 65 个低甲基化 (调整后的 P < 0.05)。前 10 个肿瘤特异性高甲基化区域中的 6 个与癌症发展有关。然后,我们比较了 TUM、ADJ 和 DIS,以进一步确定在癌症发生和早期发展过程中逐渐加重的异常甲基化。共鉴定出 332 个 DMR,其中包括一个主要比例的 312 个区域,其甲基化水平随着样本靠近肿瘤而逐渐增加 (即 DIS < ADJ < TUM),以及 20 个区域呈现出逐渐减少的模式。KEGG 和 GO 术语的基因集富集分析 (GSEA) 一致表明,位于转录因子基因中的 DMR 富集,表明转录因子的表观遗传调控在 FC 和肿瘤发生中起核心作用。
我们揭示了癌前病变和恶性肿瘤之间的不同甲基化模式,表明 DNA 甲基化作为癌前缺陷的早期步骤的重要作用。此外,我们的研究还鉴定了差异甲基化基因,特别是转录因子,它们可能被用作肺癌筛查的标志物,并用于 FC 和早期癌症发展的机制研究。