Center for Evidence-Based and Translational Medicine, Zhongnan Hospital of Wuhan University, Wuhan, 430071, China.
Department of Gastrointestinal Surgery, Huaihe Hospital of Henan University, Kaifeng, 475000, Henan, China.
Mil Med Res. 2022 Mar 28;9(1):12. doi: 10.1186/s40779-022-00373-4.
Studies had shown many diseases affect the stability of human microbiota, but how this relates to benign prostatic hyperplasia (BPH) has not been well understood. Hence, this study aimed to investigate the regulation of BPH on gut microbiota composition and metabonomics.
We analyzed gut samples from rats with BPH and healthy control rats, the gut microbiota composition and metabonomics were detected by 16S rDNA sequencing and liquid chromatography tandem mass spectrometry (LC-MS/MS).
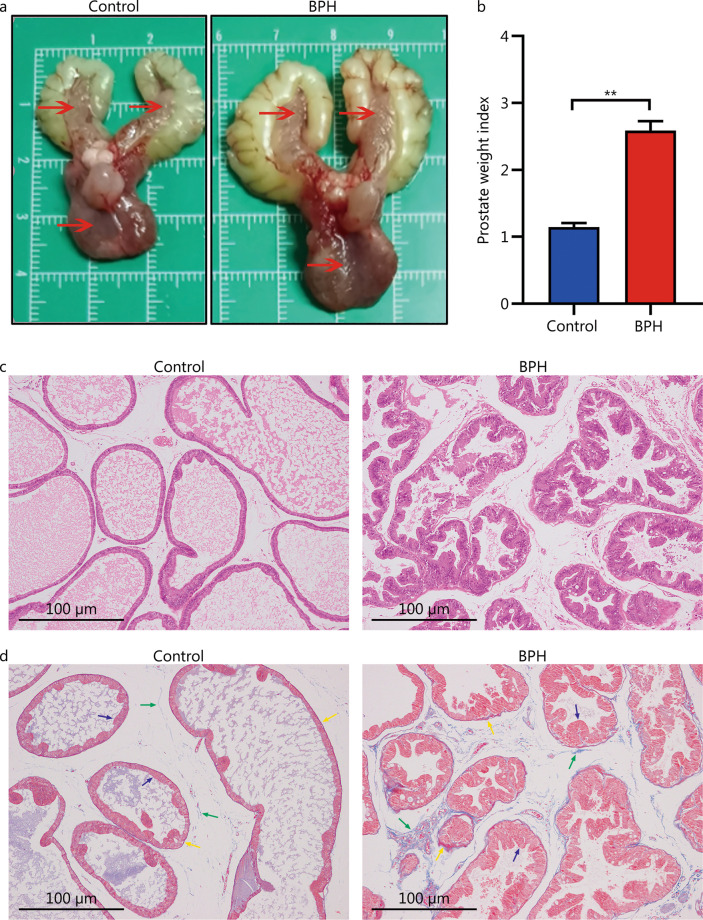
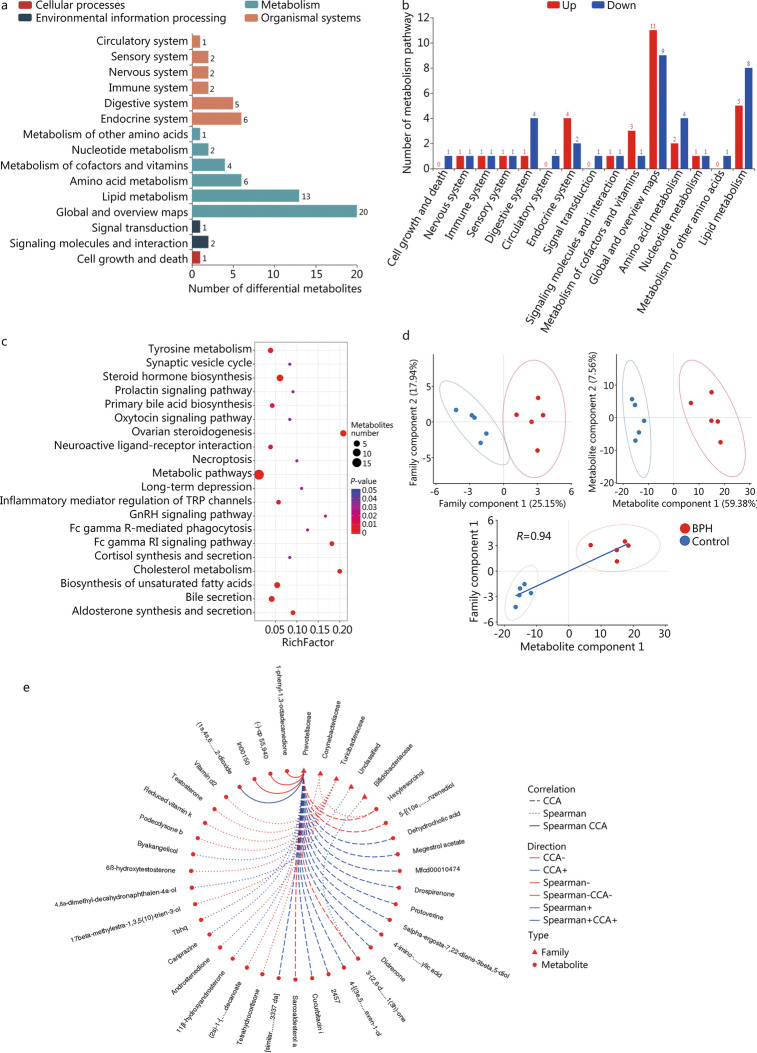
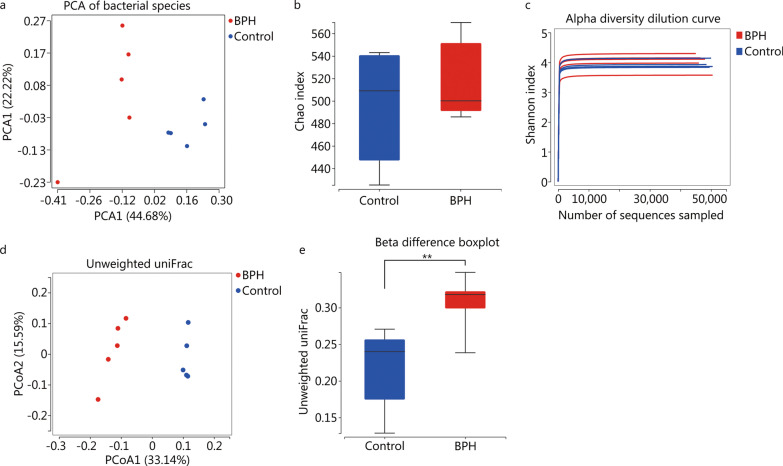
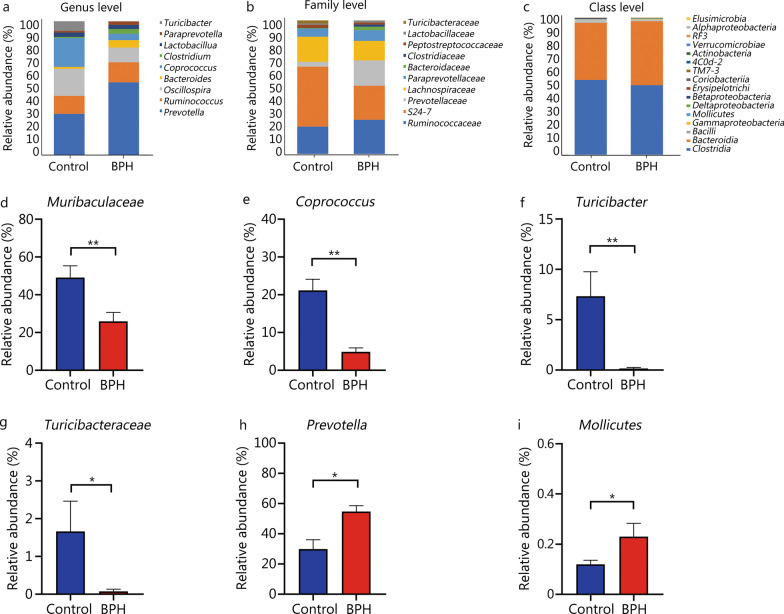
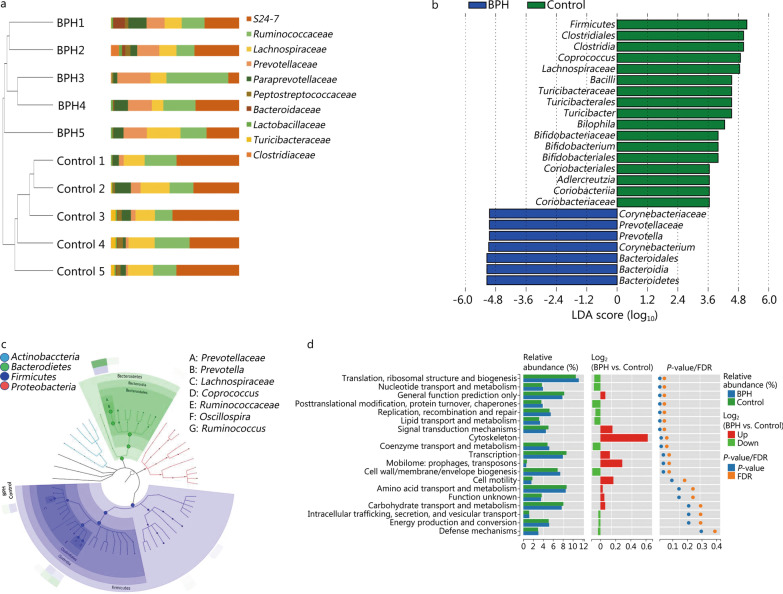
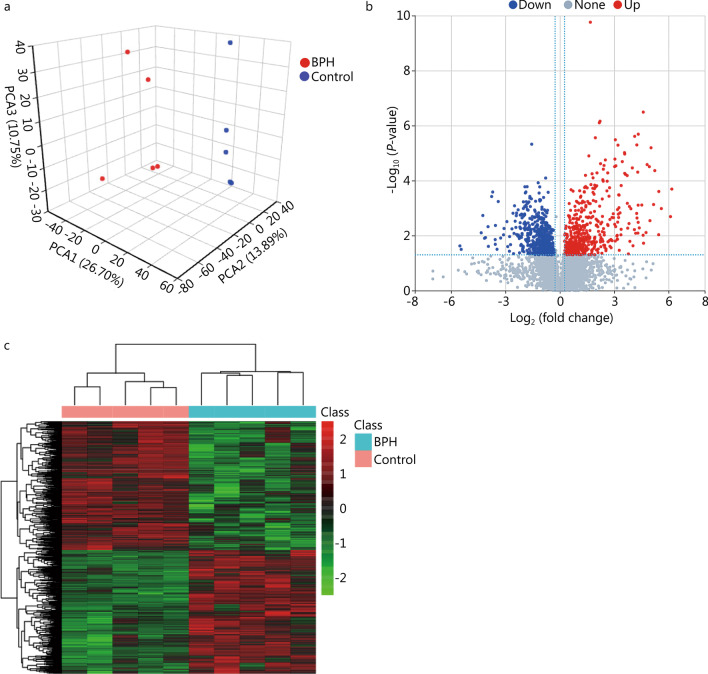
High-throughput sequencing results showed that gut microbiota beta-diversity increased (P < 0.01) in the BPH group vs. control group. Muribaculaceae (P < 0.01), Turicibacteraceae (P < 0.05), Turicibacter (P < 0.01) and Coprococcus (P < 0.01) were significantly decreased in the BPH group, whereas that of Mollicutes (P < 0.05) and Prevotella (P < 0.05) were significantly increased compared with the control group. Despite profound interindividual variability, the levels of several predominant genera were different. In addition, there were no statistically significant differences in several bacteria. BPH group vs. control group: Firmicutes (52.30% vs. 57.29%, P > 0.05), Bacteroidetes (46.54% vs. 41.64%, P > 0.05), Clostridia (50.89% vs. 54.66%, P > 0.05), Ruminococcaceae (25.67% vs. 20.56%, P > 0.05). LC-MS/MS of intestinal contents revealed that differential metabolites were mainly involved in cellular processes, environmental information processing, metabolism and organismal systems. The most important pathways were global and overview maps, lipid metabolism, amino acid metabolism, digestive system and endocrine system. Through enrichment analysis, we found that the differential metabolites were significantly enriched in metabolic pathways, steroid hormone biosynthesis, ovarian steroidogenesis, biosynthesis of unsaturated fatty acids and bile secretion. Pearson correlation analysis (R = 0.94) showed that there was a strong correlation between Prevotellaceae, Corynebacteriaceae, Turicibacteraceae, Bifidobacteriaceae and differential metabolites.
Our findings suggested an association between the gut microbiota and BPH, but the causal relationship between the two groups is unclear. Thus, further studies are warranted to elucidate the potential mechanisms and causal relationships between BPH and gut microbiota.
研究表明,许多疾病会影响人体微生物群的稳定性,但这与良性前列腺增生(BPH)的关系尚不清楚。因此,本研究旨在探讨 BPH 对肠道微生物群组成和代谢组学的调节作用。
我们分析了 BPH 大鼠和健康对照组大鼠的肠道样本,通过 16S rDNA 测序和液相色谱串联质谱(LC-MS/MS)检测肠道微生物群组成和代谢组学。
高通量测序结果显示,BPH 组肠道微生物群β多样性增加(P<0.01)。与对照组相比,BPH 组中的 Muribaculaceae(P<0.01)、Turicibacteraceae(P<0.05)、Turicibacter(P<0.01)和 Coprococcus(P<0.01)明显减少,而 Mollicutes(P<0.05)和 Prevotella(P<0.05)明显增加。尽管个体间存在明显的变异性,但几个主要属的水平有所不同。此外,还有一些细菌没有统计学意义上的差异。BPH 组与对照组相比:厚壁菌门(52.30%对 57.29%,P>0.05),拟杆菌门(46.54%对 41.64%,P>0.05),梭菌属(50.89%对 54.66%,P>0.05),瘤胃球菌科(25.67%对 20.56%,P>0.05)。肠道内容物的 LC-MS/MS 分析显示,差异代谢物主要涉及细胞过程、环境信息处理、代谢和机体系统。最重要的途径是全局和概述图、脂质代谢、氨基酸代谢、消化系统和内分泌系统。通过富集分析,我们发现差异代谢物在代谢途径、甾体激素生物合成、卵巢类固醇生成、不饱和脂肪酸合成和胆汁分泌中显著富集。Pearson 相关分析(R=0.94)显示,普雷沃氏菌科、棒状杆菌科、瘤胃球菌科、双歧杆菌科与差异代谢物之间存在很强的相关性。
我们的研究结果提示肠道微生物群与 BPH 之间存在关联,但两者之间的因果关系尚不清楚。因此,需要进一步的研究来阐明 BPH 与肠道微生物群之间的潜在机制和因果关系。