Stanley Center for Psychiatric Research, Broad Institute of MIT and Harvard, Cambridge, MA, USA.
Bio-X Institutes, Key Laboratory for the Genetics of Developmental and Neuropsychiatric Disorders (Ministry of Education), Shanghai Jiao Tong University, Shanghai, China.
Nat Genet. 2022 May;54(5):573-580. doi: 10.1038/s41588-022-01054-7. Epub 2022 May 5.
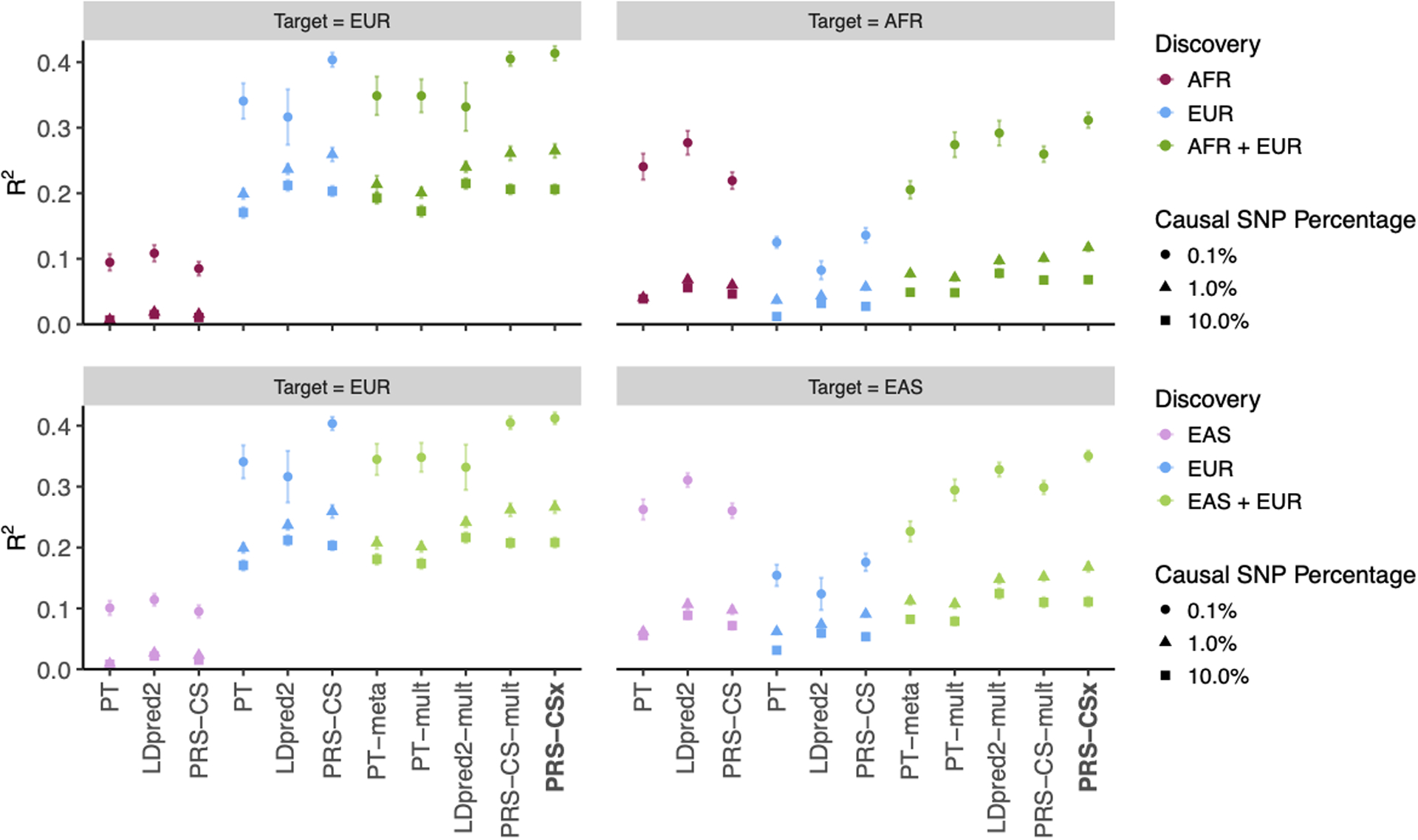
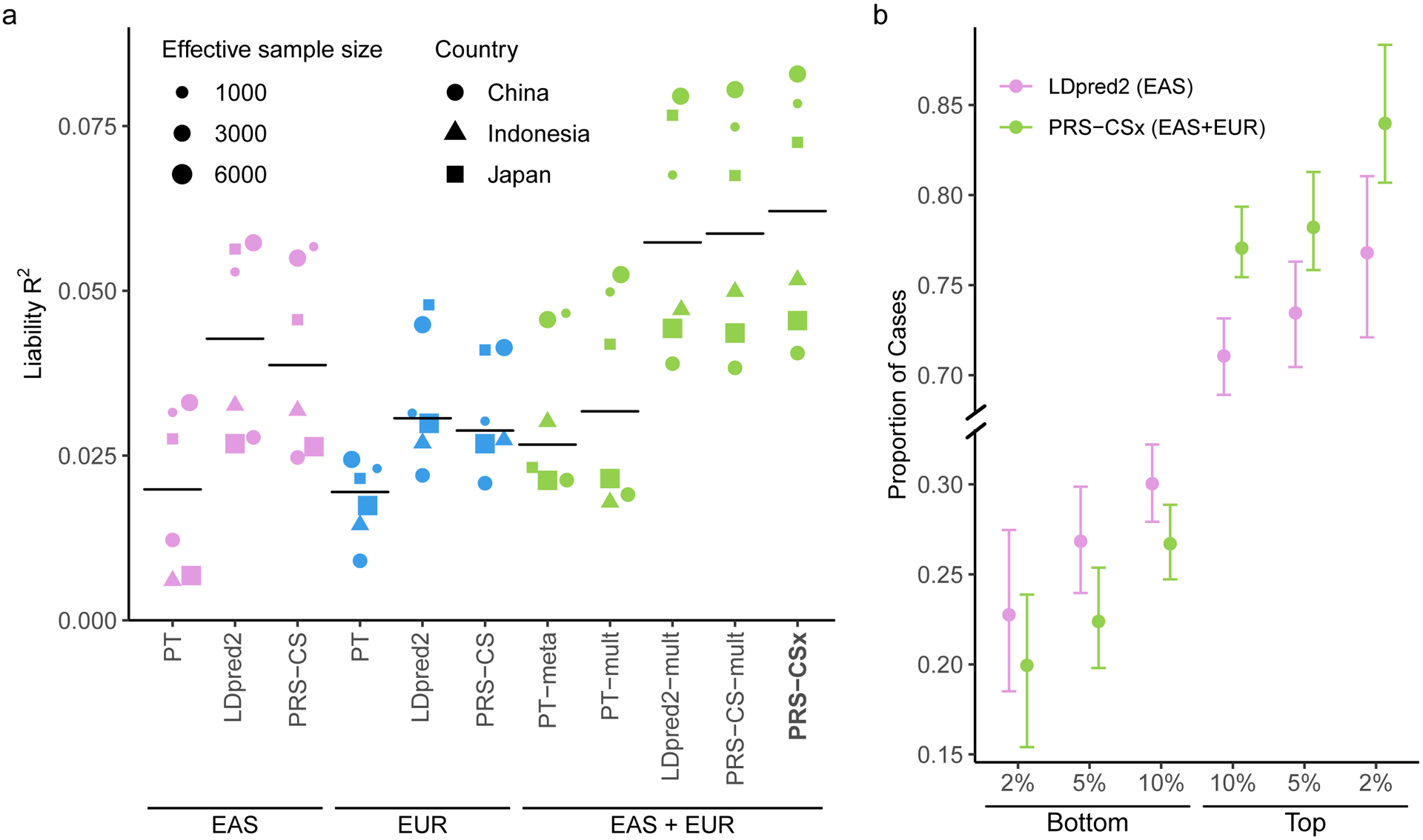
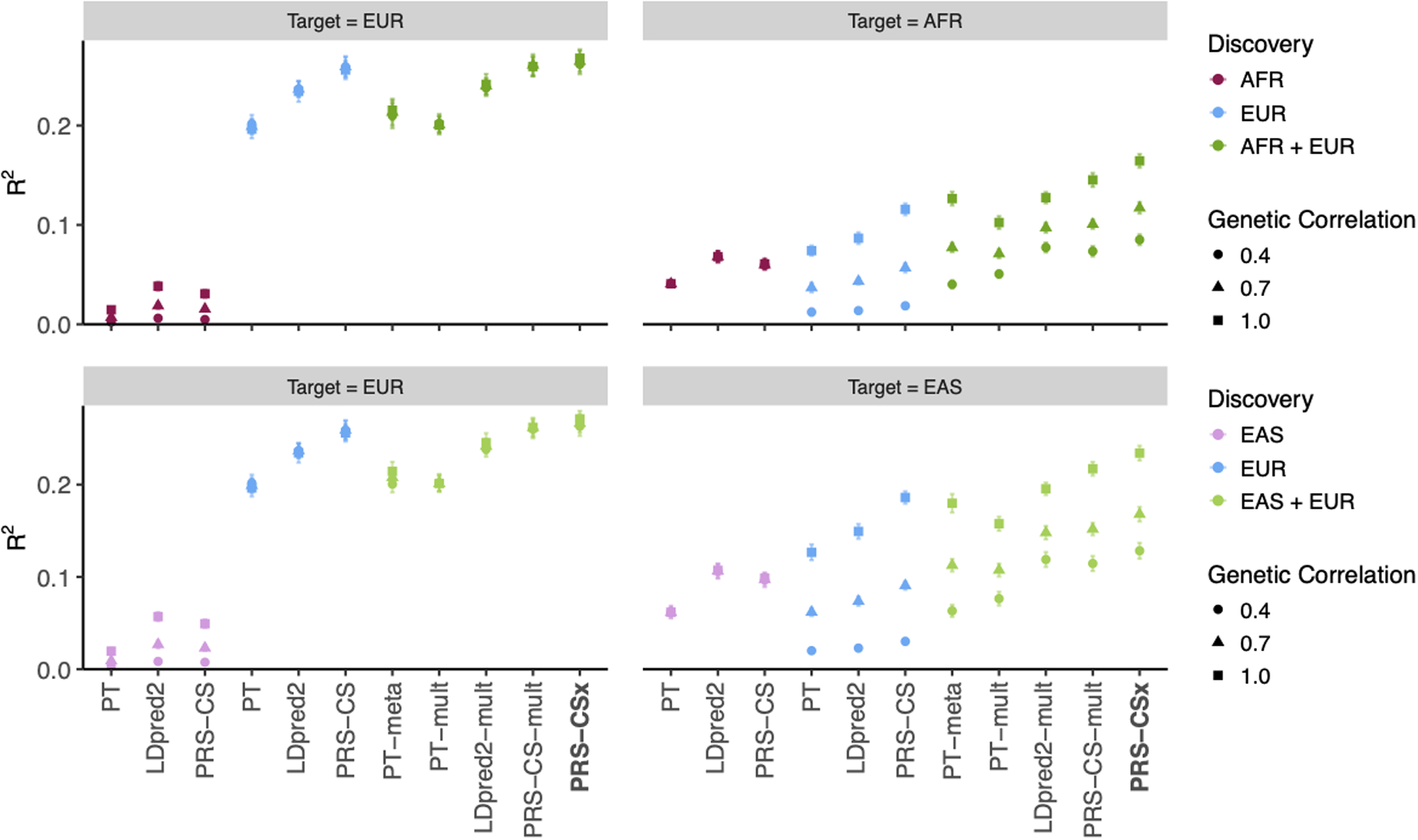
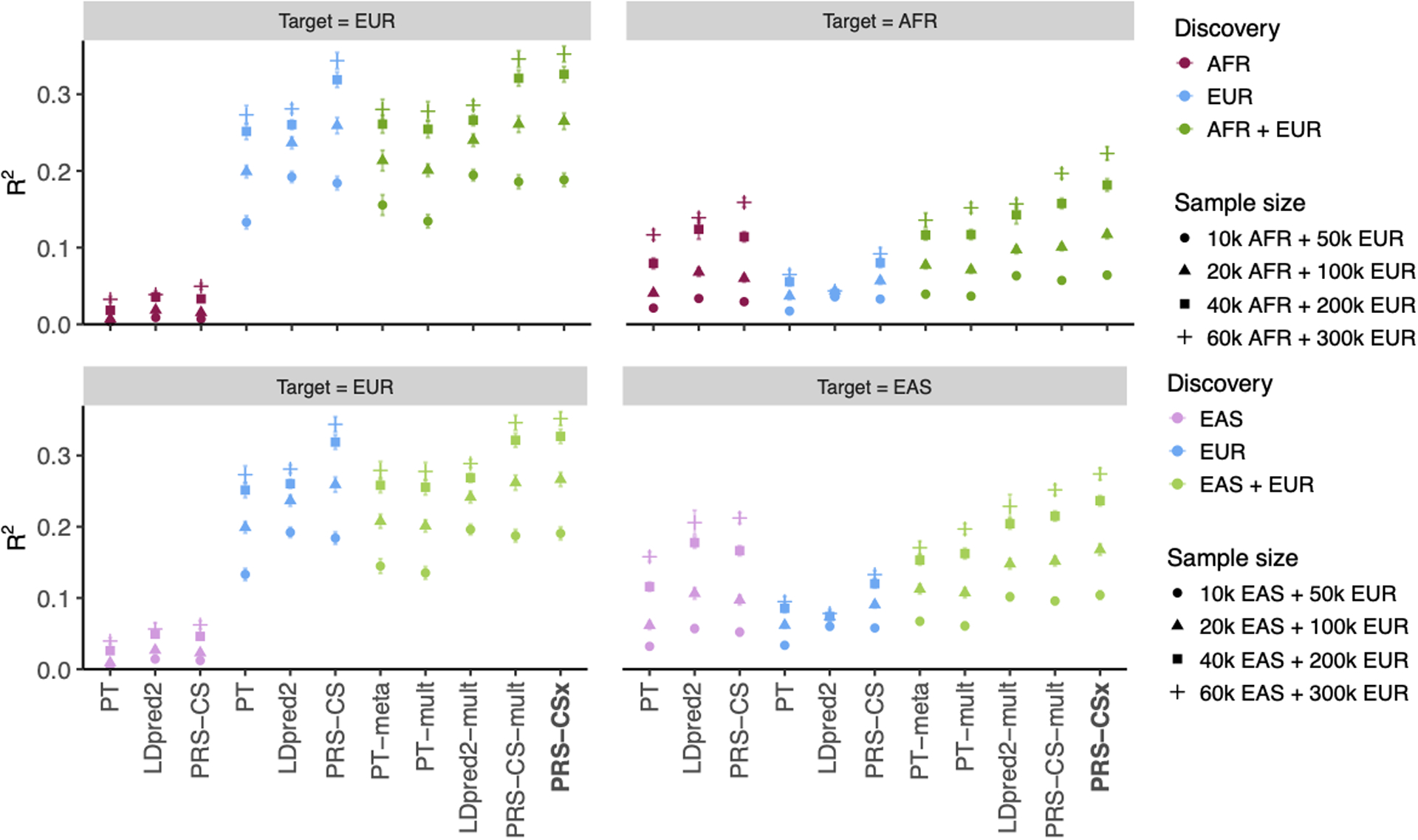
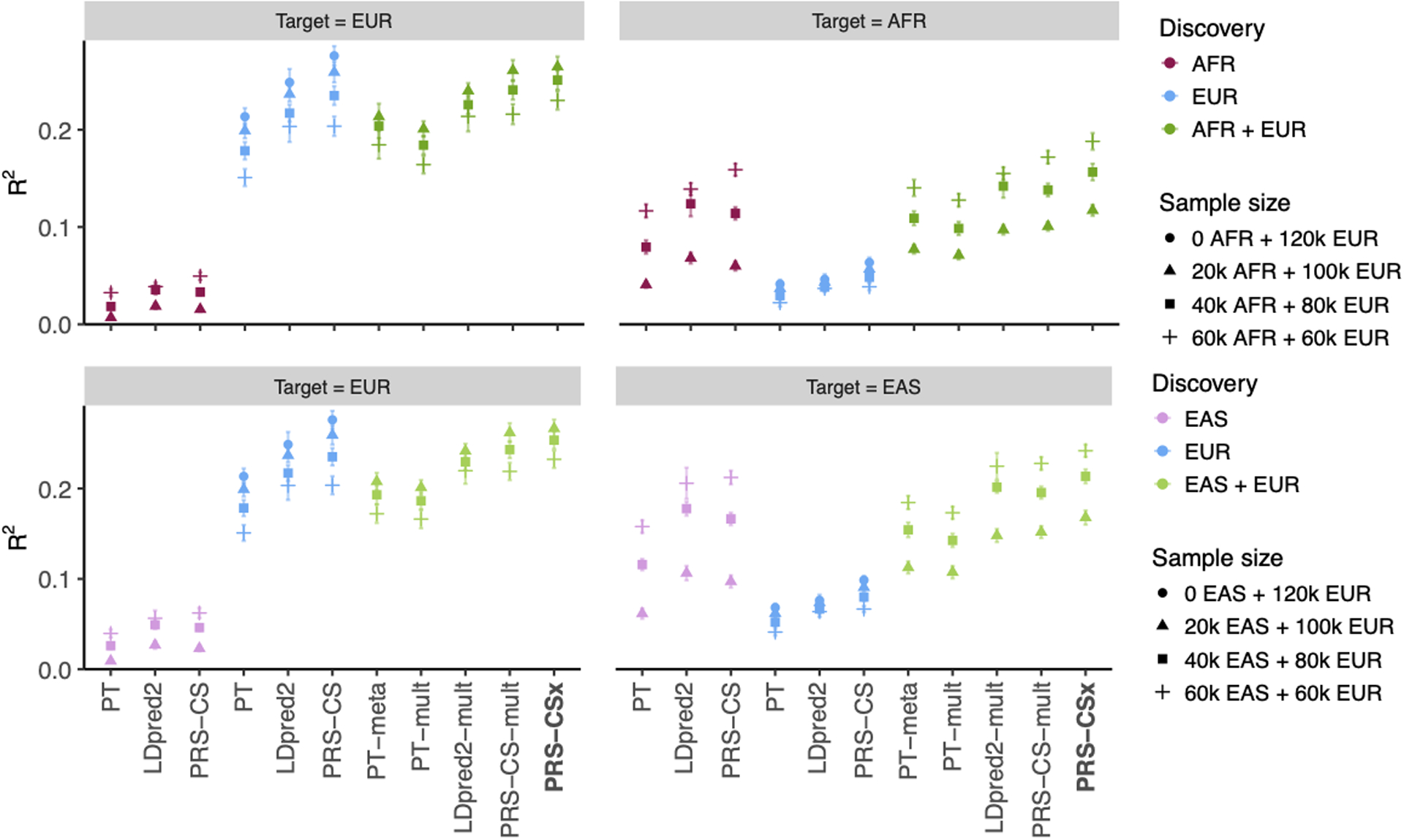
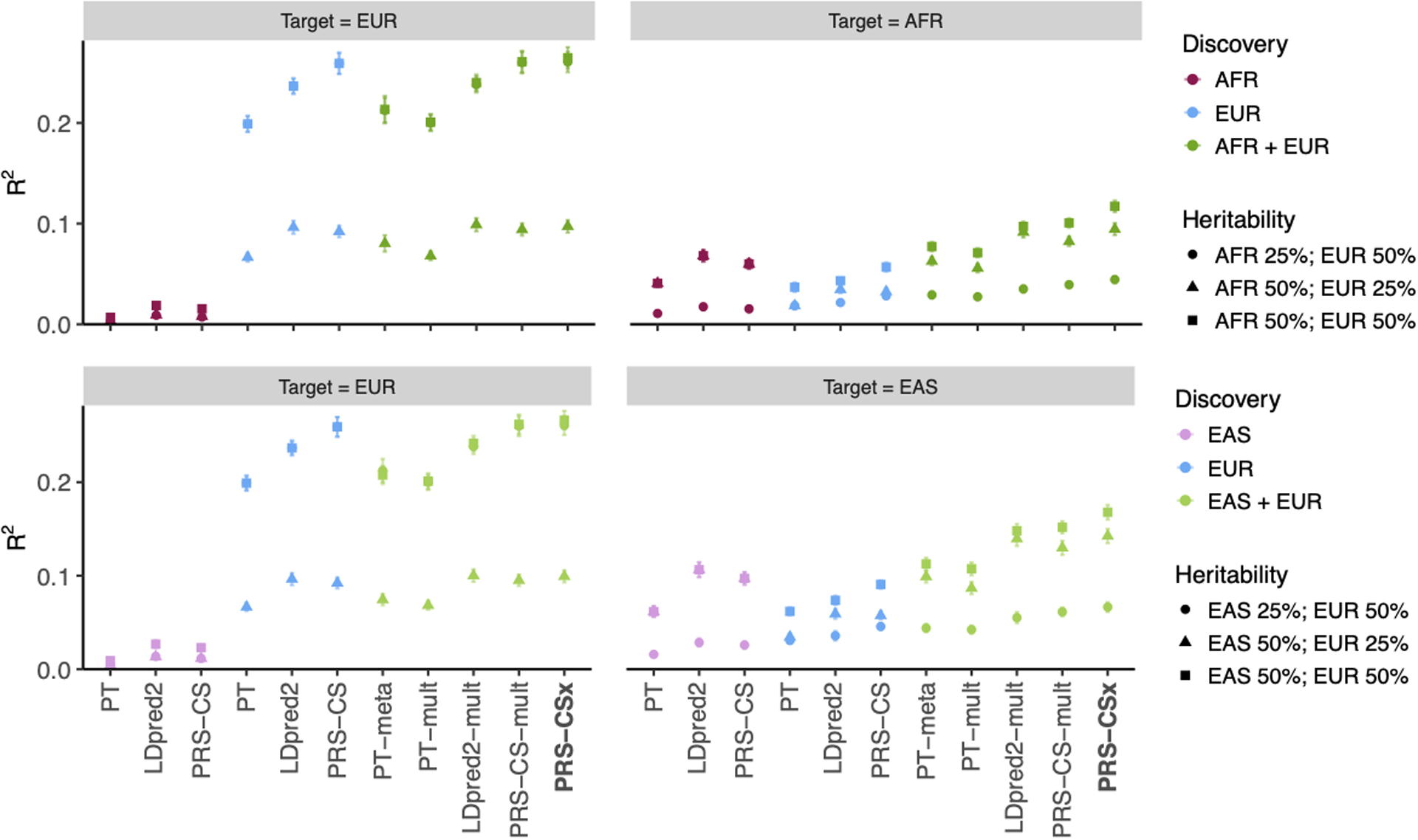
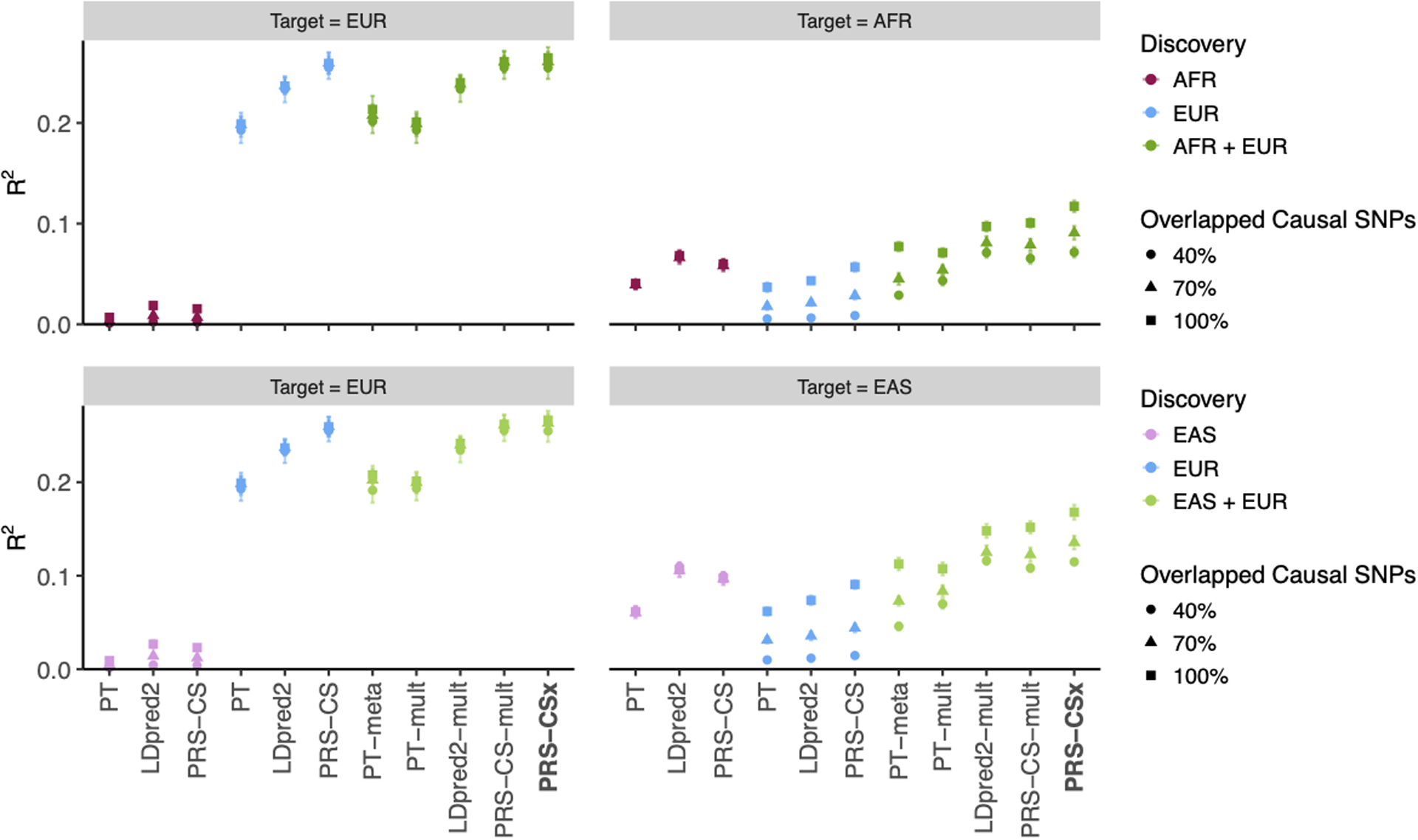
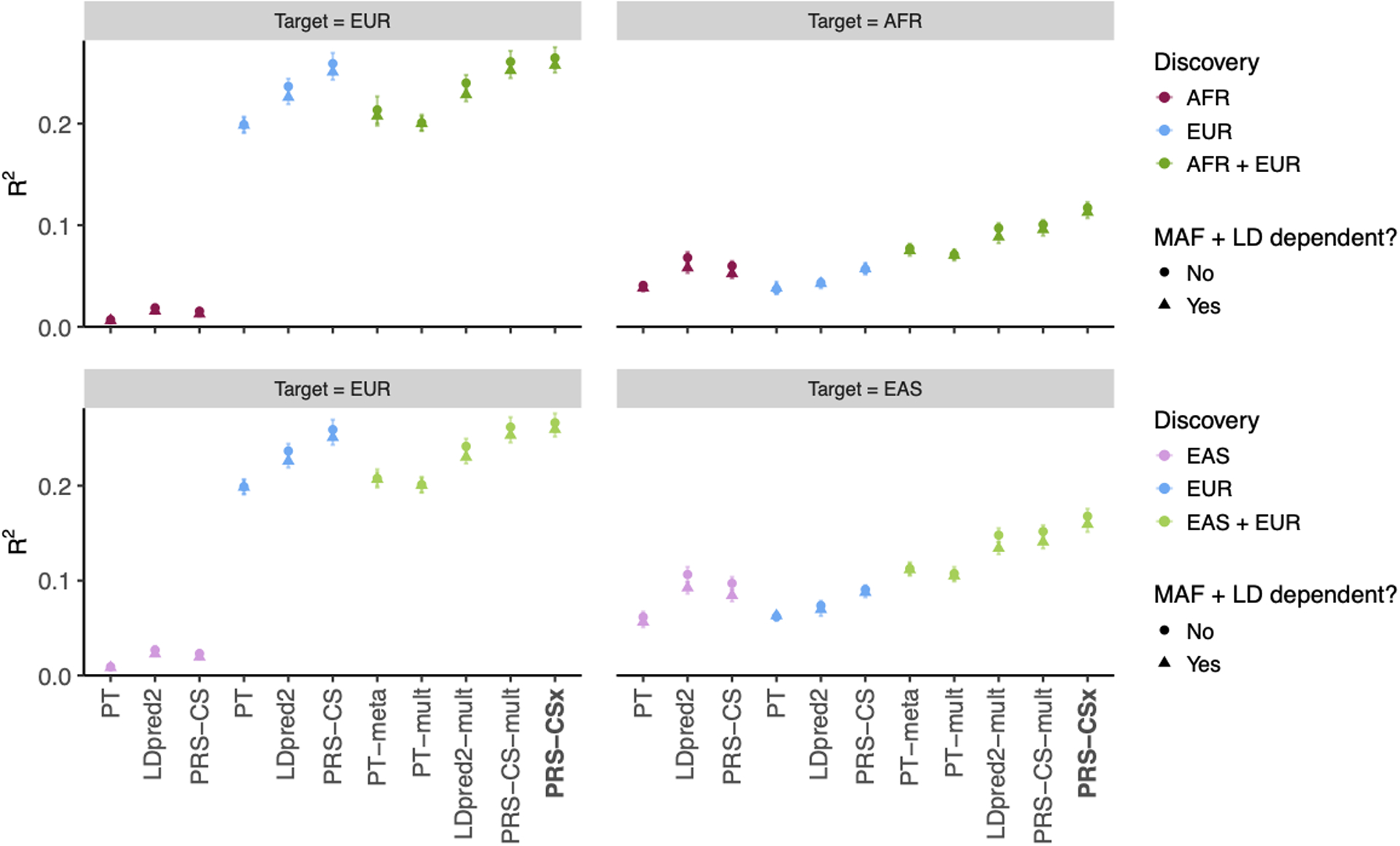
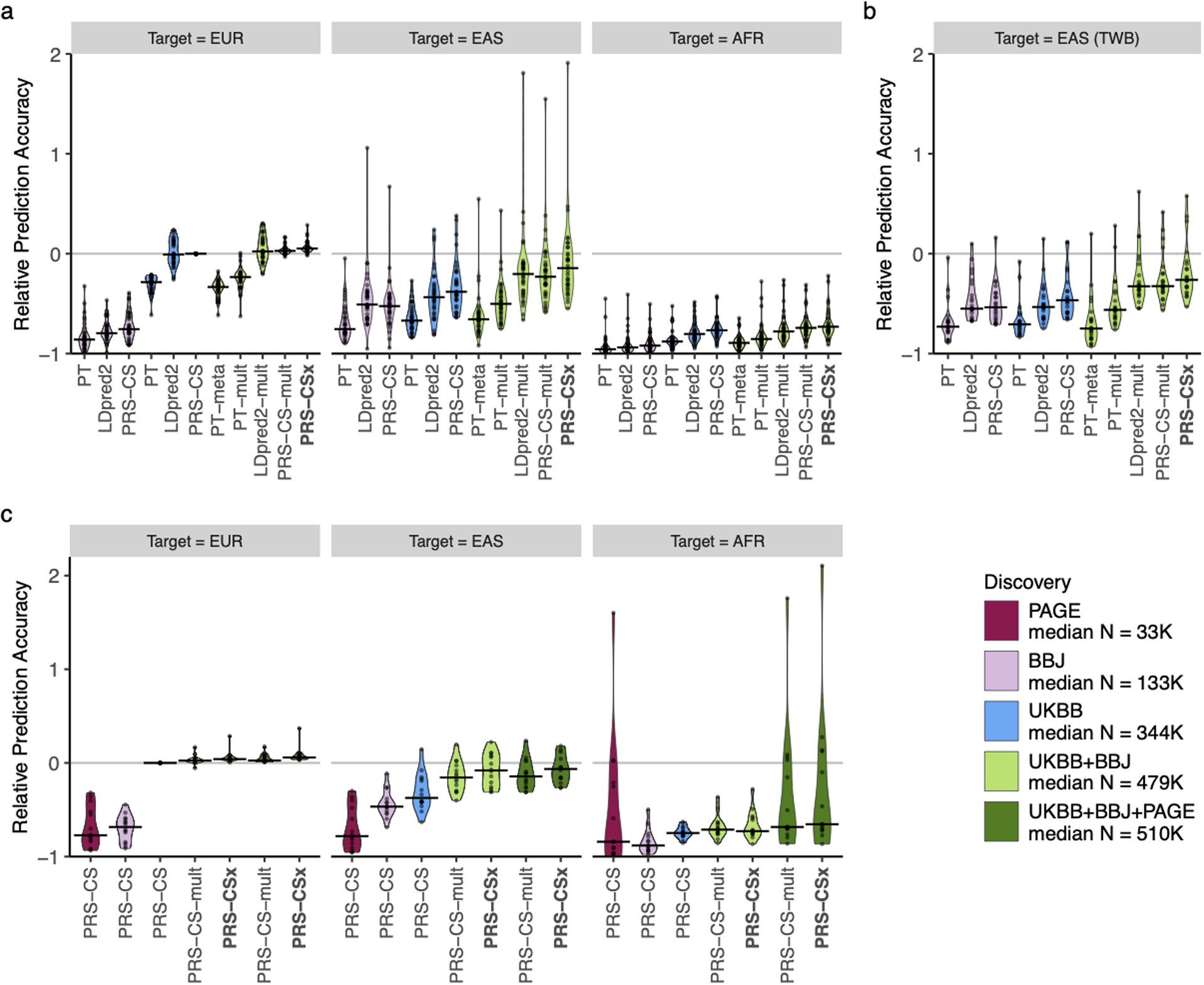
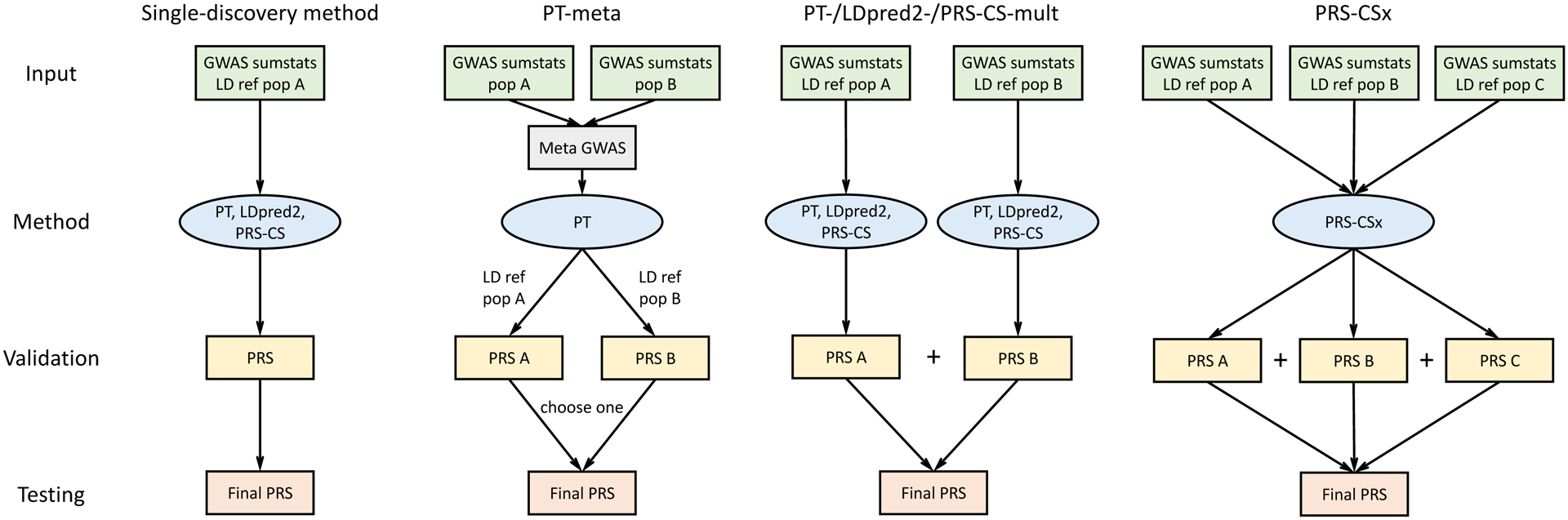
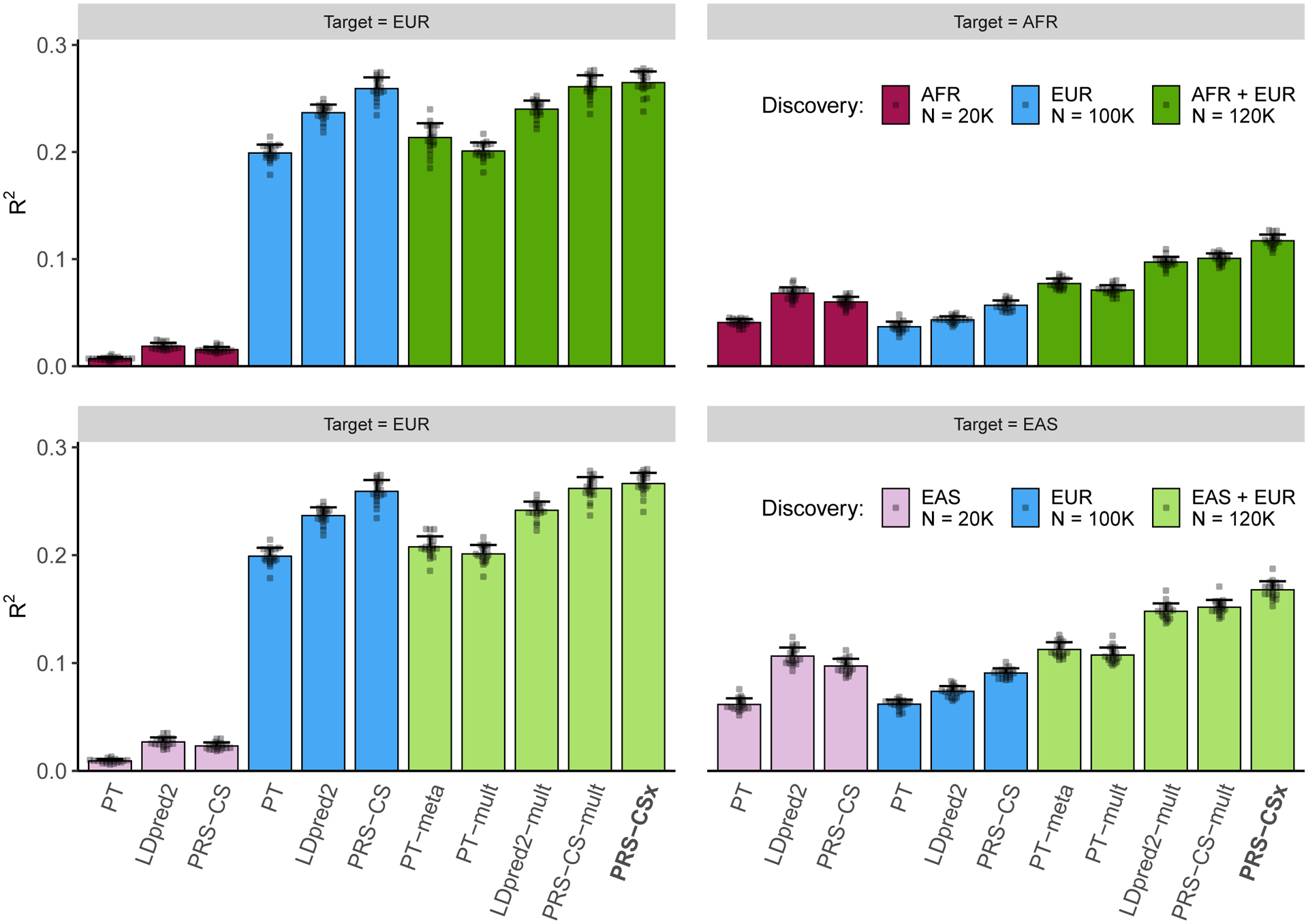
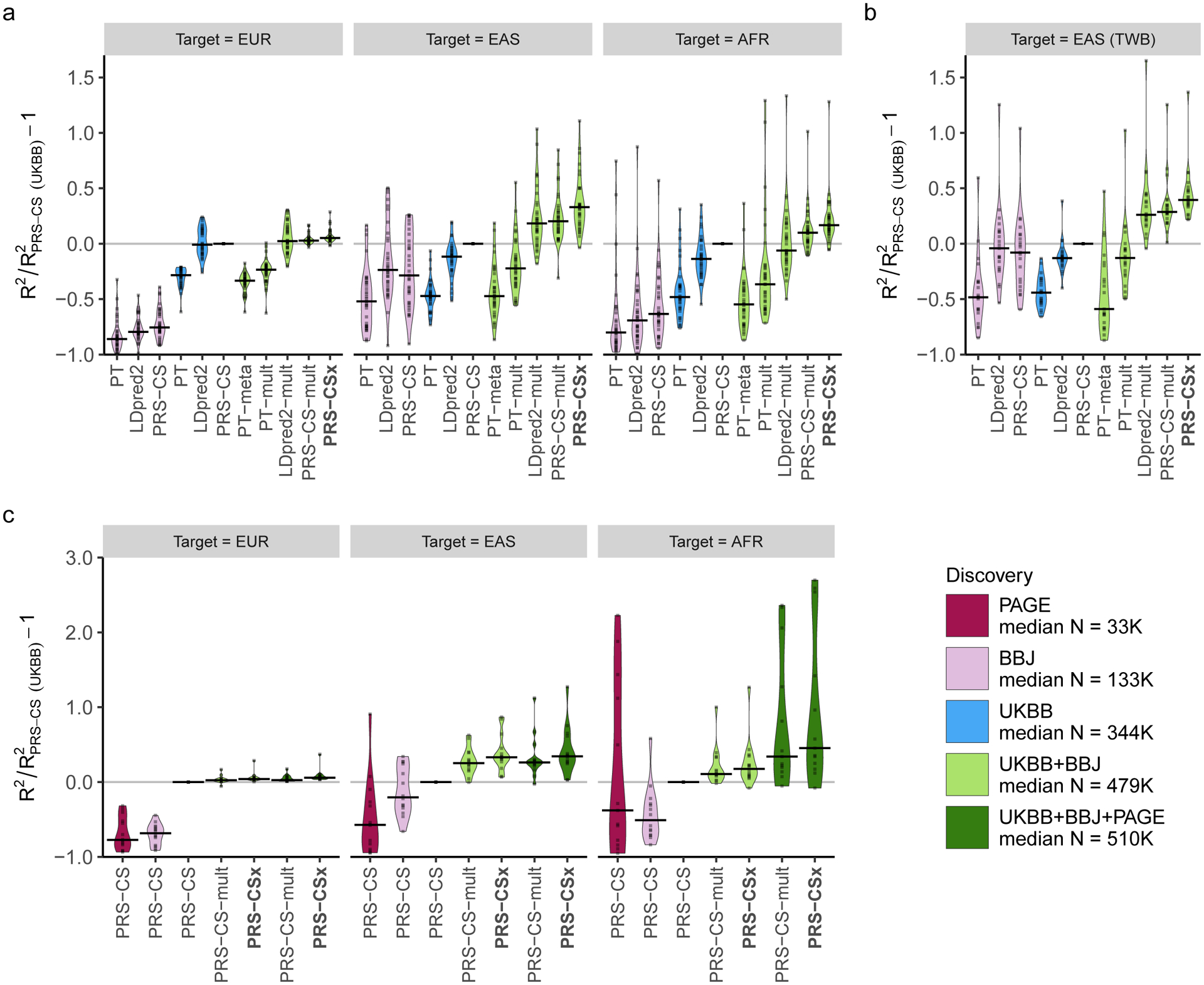
Polygenic risk scores (PRS) have attenuated cross-population predictive performance. As existing genome-wide association studies (GWAS) have been conducted predominantly in individuals of European descent, the limited transferability of PRS reduces their clinical value in non-European populations, and may exacerbate healthcare disparities. Recent efforts to level ancestry imbalance in genomic research have expanded the scale of non-European GWAS, although most remain underpowered. Here, we present a new PRS construction method, PRS-CSx, which improves cross-population polygenic prediction by integrating GWAS summary statistics from multiple populations. PRS-CSx couples genetic effects across populations via a shared continuous shrinkage (CS) prior, enabling more accurate effect size estimation by sharing information between summary statistics and leveraging linkage disequilibrium diversity across discovery samples, while inheriting computational efficiency and robustness from PRS-CS. We show that PRS-CSx outperforms alternative methods across traits with a wide range of genetic architectures, cross-population genetic overlaps and discovery GWAS sample sizes in simulations, and improves the prediction of quantitative traits and schizophrenia risk in non-European populations.
多基因风险评分(PRS)的跨人群预测性能已经减弱。由于现有的全基因组关联研究(GWAS)主要在欧洲血统的个体中进行,PRS 的有限可转移性降低了其在非欧洲人群中的临床价值,并可能加剧医疗保健差距。最近在基因组研究中努力平衡祖先背景的努力扩大了非欧洲 GWAS 的规模,尽管大多数研究仍然没有足够的效力。在这里,我们提出了一种新的 PRS 构建方法 PRS-CSx,该方法通过整合来自多个群体的 GWAS 汇总统计数据来提高跨人群多基因预测的能力。PRS-CSx 通过共享连续收缩(CS)先验来跨人群耦合遗传效应,通过在汇总统计数据之间共享信息并利用发现样本中的连锁不平衡多样性来实现更准确的效应大小估计,同时继承了 PRS-CS 的计算效率和稳健性。我们在模拟中表明,PRS-CSx 在具有广泛遗传结构、跨人群遗传重叠和发现 GWAS 样本大小的多种性状上的表现优于其他方法,并提高了非欧洲人群中数量性状和精神分裂症风险的预测能力。