Department of Biostatistics and Medical Informatics, University of Wisconsin-Madison, Madison, WI, 53706, USA.
Center for Statistical Science, Department of Industrial Engineering, Tsinghua University, Beijing, 100084, China.
Nat Commun. 2023 Feb 14;14(1):832. doi: 10.1038/s41467-023-36544-7.
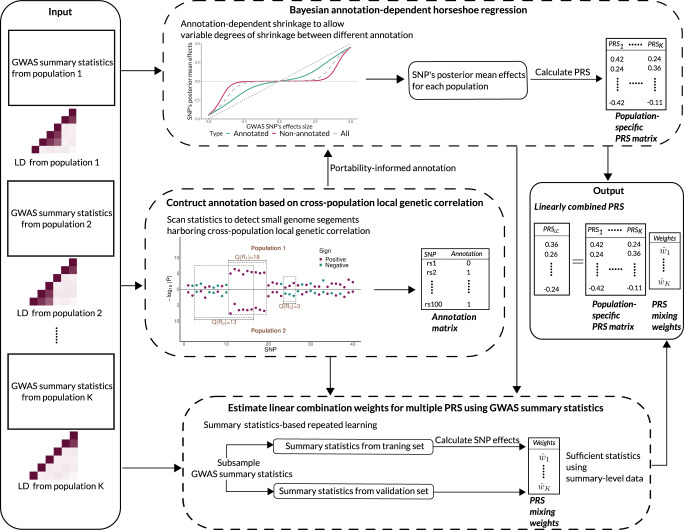
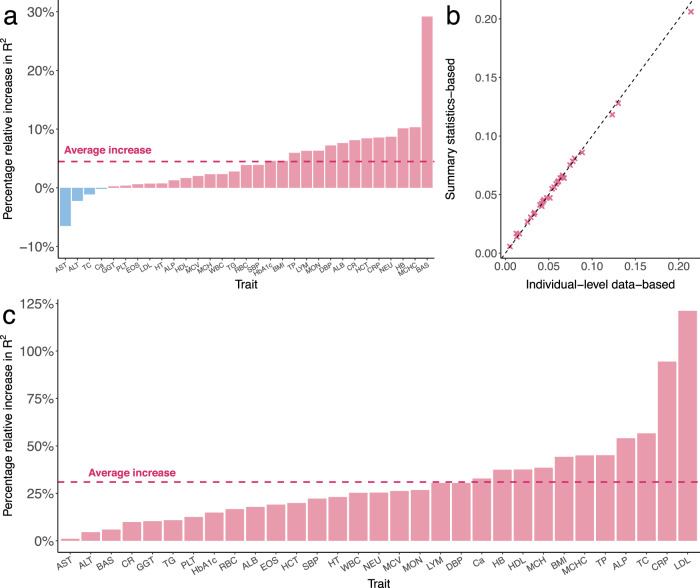
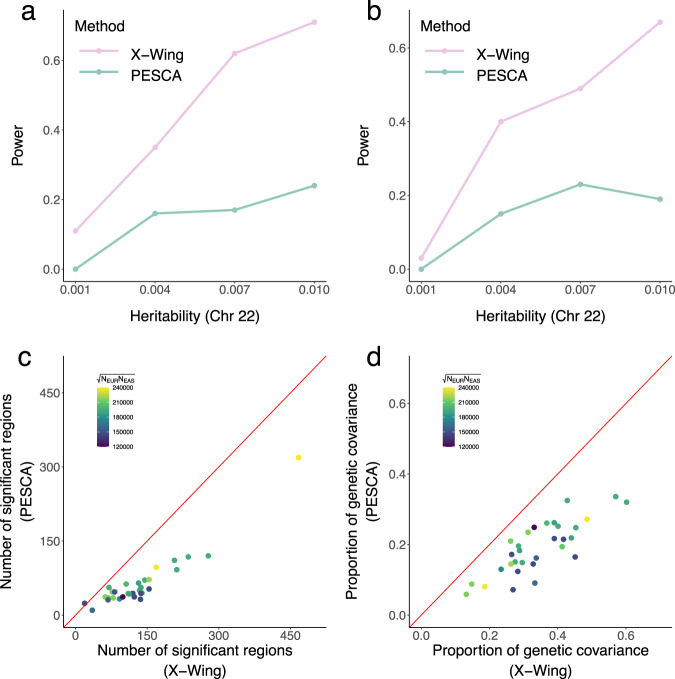
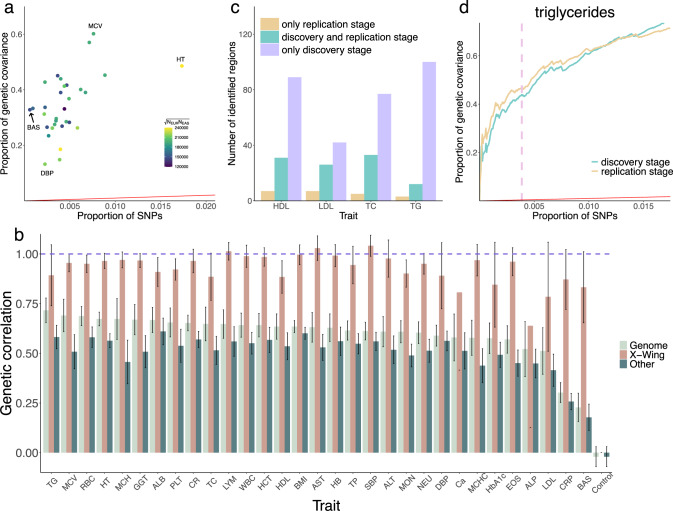
Polygenic risk scores (PRS) calculated from genome-wide association studies (GWAS) of Europeans are known to have substantially reduced predictive accuracy in non-European populations, limiting their clinical utility and raising concerns about health disparities across ancestral populations. Here, we introduce a statistical framework named X-Wing to improve predictive performance in ancestrally diverse populations. X-Wing quantifies local genetic correlations for complex traits between populations, employs an annotation-dependent estimation procedure to amplify correlated genetic effects between populations, and combines multiple population-specific PRS into a unified score with GWAS summary statistics alone as input. Through extensive benchmarking, we demonstrate that X-Wing pinpoints portable genetic effects and substantially improves PRS performance in non-European populations, showing 14.1%-119.1% relative gain in predictive R compared to state-of-the-art methods based on GWAS summary statistics. Overall, X-Wing addresses critical limitations in existing approaches and may have broad applications in cross-population polygenic risk prediction.
基于全基因组关联研究(GWAS)计算的多基因风险评分(PRS)在非欧洲人群中的预测准确性显著降低,限制了其临床应用,并引发了对不同祖先人群健康差异的关注。在这里,我们引入了一种名为 X-Wing 的统计框架,以提高在具有多种祖先人群中的预测性能。X-Wing 量化了不同人群之间复杂性状的局部遗传相关性,采用依赖注释的估计程序来放大人群之间相关的遗传效应,并将多个特定人群的 PRS 结合起来,仅使用 GWAS 汇总统计数据作为输入,形成一个统一的评分。通过广泛的基准测试,我们证明了 X-Wing 可以确定可移植的遗传效应,并显著提高非欧洲人群中的 PRS 性能,与基于 GWAS 汇总统计数据的最先进方法相比,预测 R 的相对增益为 14.1%-119.1%。总体而言,X-Wing 解决了现有方法的关键局限性,并且可能在跨人群多基因风险预测中有广泛的应用。