Verhellen Jonas
Centre for Integrative Neuroplasticity, University of Oslo N-0316 Oslo Norway
Chem Sci. 2022 Jun 2;13(25):7526-7535. doi: 10.1039/d2sc00821a. eCollection 2022 Jun 29.
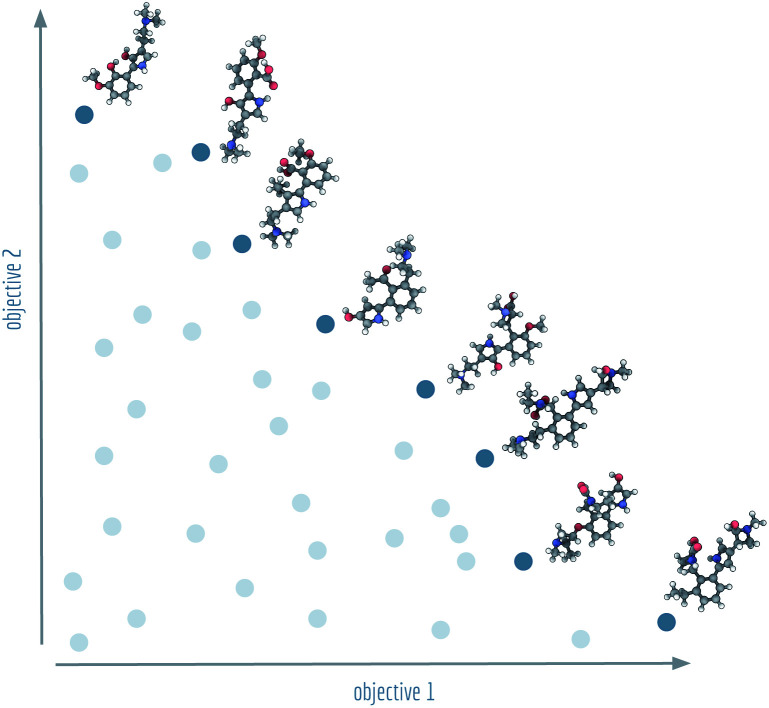
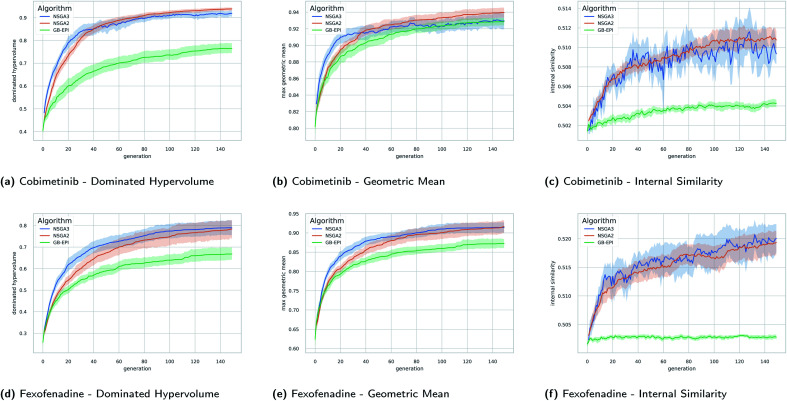
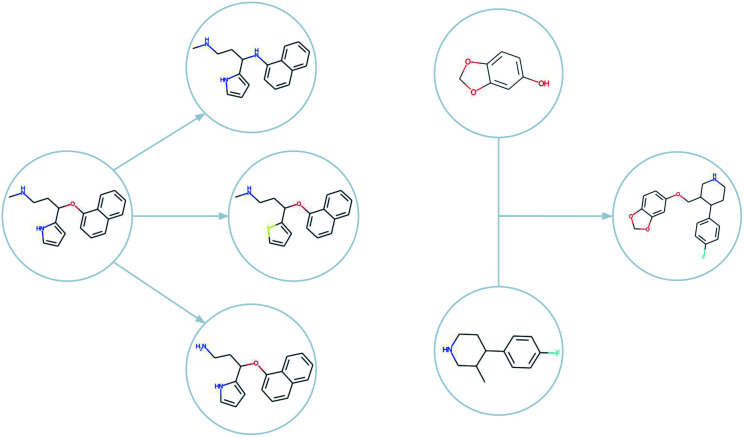
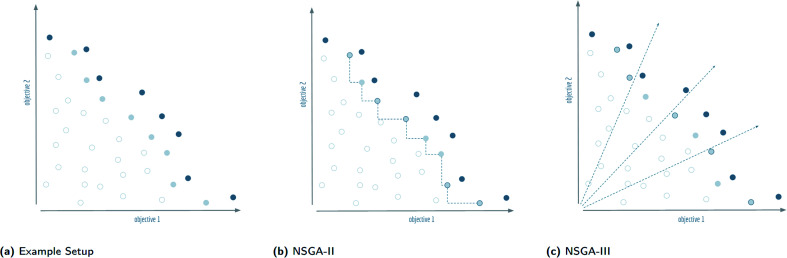
Computer-assisted design of small molecules has experienced a resurgence in academic and industrial interest due to the widespread use of data-driven techniques such as deep generative models. While the ability to generate molecules that fulfil required chemical properties is encouraging, the use of deep learning models requires significant, if not prohibitive, amounts of data and computational power. At the same time, open-sourcing of more traditional techniques such as graph-based genetic algorithms for molecular optimisation [Jensen, , 2019, , 3567-3572] has shown that simple and training-free algorithms can be efficient and robust alternatives. Further research alleviated the common genetic algorithm issue of evolutionary stagnation by enforcing molecular diversity during optimisation [Van den Abeele, , 2020, , 11485-11491]. The crucial lesson distilled from the simultaneous development of deep generative models and advanced genetic algorithms has been the importance of chemical space exploration [Aspuru-Guzik, , 2021, , 7079-7090]. For single-objective optimisation problems, chemical space exploration had to be discovered as a useable resource but in multi-objective optimisation problems, an exploration of trade-offs between conflicting objectives is inherently present. In this paper we provide state-of-the-art and open-source implementations of two generations of graph-based non-dominated sorting genetic algorithms (NSGA-II, NSGA-III) for molecular multi-objective optimisation. We provide the results of a series of benchmarks for the inverse design of small molecule drugs for both the NSGA-II and NSGA-III algorithms. In addition, we introduce the dominated hypervolume and extended fingerprint based internal similarity as novel metrics for these benchmarks. By design, NSGA-II, and NSGA-III outperform a single optimisation method baseline in terms of dominated hypervolume, but remarkably our results show they do so without relying on a greater internal chemical diversity.
由于深度生成模型等数据驱动技术的广泛应用,小分子的计算机辅助设计在学术界和工业界重新引起了关注。虽然生成具有所需化学性质的分子的能力令人鼓舞,但深度学习模型的使用需要大量(即便不是高得令人望而却步)的数据和计算能力。与此同时,分子优化的更传统技术(如基于图的遗传算法)的开源 [詹森,2019,3567 - 3572] 表明,简单且无需训练的算法可以是高效且稳健的替代方案。进一步的研究通过在优化过程中强制分子多样性缓解了遗传算法常见的进化停滞问题 [范登·阿贝勒,2020,11485 - 11491]。从深度生成模型和先进遗传算法的同步发展中汲取的关键教训是化学空间探索的重要性 [阿斯普鲁 - 古齐克,2021,7079 - 7090]。对于单目标优化问题,化学空间探索必须作为一种可用资源被发现,但在多目标优化问题中,冲突目标之间权衡的探索是内在存在的。在本文中,我们提供了两代基于图的非支配排序遗传算法(NSGA - II、NSGA - III)用于分子多目标优化的最新开源实现。我们给出了针对NSGA - II和NSGA - III算法的小分子药物逆设计的一系列基准测试结果。此外,我们引入支配超体积和基于扩展指纹的内部相似度作为这些基准测试的新指标。从设计角度来看,NSGA - II和NSGA - III在支配超体积方面优于单一优化方法基线,但值得注意的是,我们的结果表明它们这样做并不依赖于更大的内部化学多样性。