Scientific Computing and Research Support Unit, University of Lausanne, 1015 Lausanne, Switzerland.
Department of Quantitative Biomedicine, University of Zurich, 8057 Zurich, Switzerland.
Proc Natl Acad Sci U S A. 2022 Aug 2;119(31):e2121279119. doi: 10.1073/pnas.2121279119. Epub 2022 Jul 29.
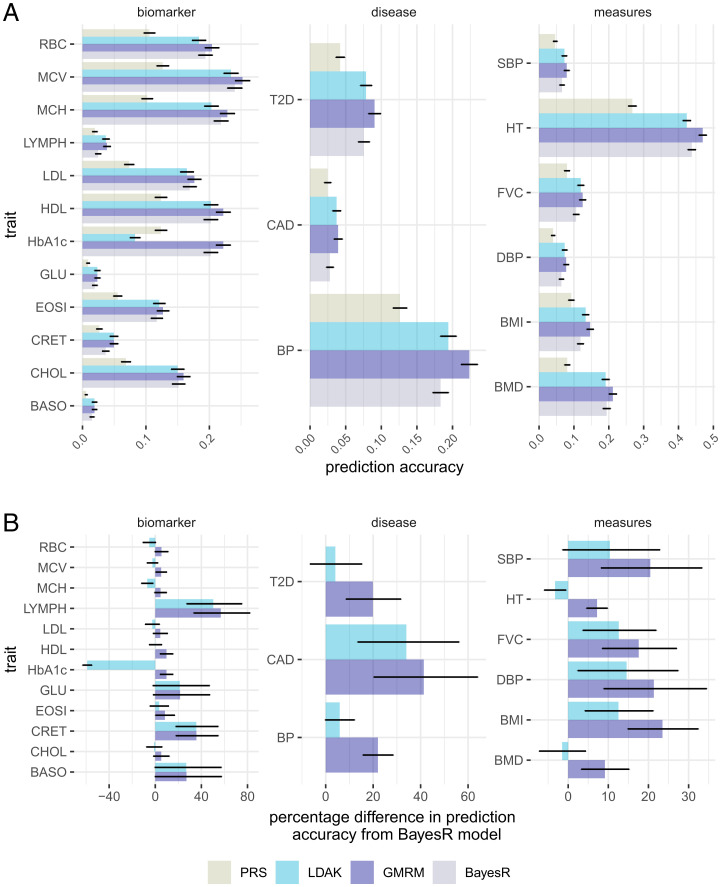
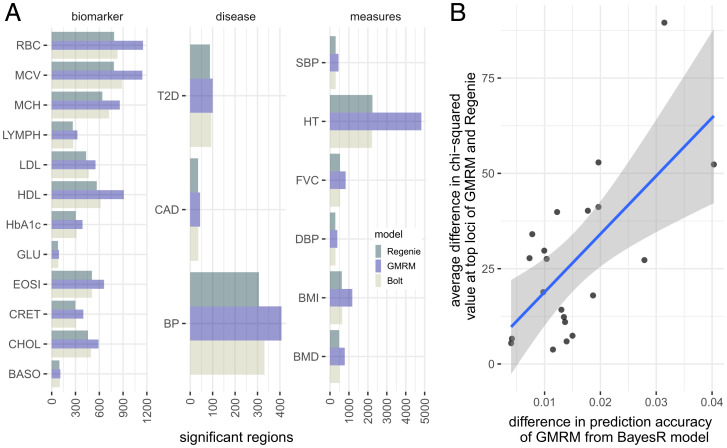
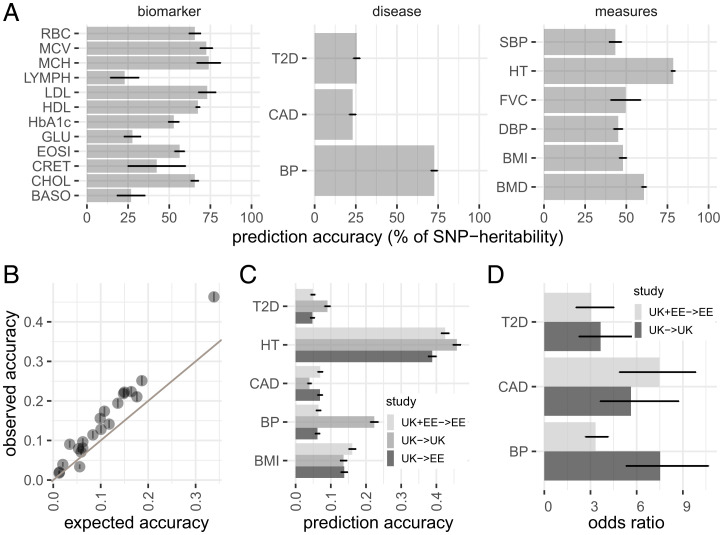
Genetically informed, deep-phenotyped biobanks are an important research resource and it is imperative that the most powerful, versatile, and efficient analysis approaches are used. Here, we apply our recently developed Bayesian grouped mixture of regressions model (GMRM) in the UK and Estonian Biobanks and obtain the highest genomic prediction accuracy reported to date across 21 heritable traits. When compared to other approaches, GMRM accuracy was greater than annotation prediction models run in the LDAK or LDPred-funct software by 15% (SE 7%) and 14% (SE 2%), respectively, and was 18% (SE 3%) greater than a baseline BayesR model without single-nucleotide polymorphism (SNP) markers grouped into minor allele frequency-linkage disequilibrium (MAF-LD) annotation categories. For height, the prediction accuracy was 47% in a UK Biobank holdout sample, which was 76% of the estimated [Formula: see text]. We then extend our GMRM prediction model to provide mixed-linear model association (MLMA) SNP marker estimates for genome-wide association (GWAS) discovery, which increased the independent loci detected to 16,162 in unrelated UK Biobank individuals, compared to 10,550 from BoltLMM and 10,095 from Regenie, a 62 and 65% increase, respectively. The average [Formula: see text] value of the leading markers increased by 15.24 (SE 0.41) for every 1% increase in prediction accuracy gained over a baseline BayesR model across the traits. Thus, we show that modeling genetic associations accounting for MAF and LD differences among SNP markers, and incorporating prior knowledge of genomic function, is important for both genomic prediction and discovery in large-scale individual-level studies.
基于遗传信息和深度表型的生物库是一项重要的研究资源,因此必须使用最强大、最通用和最高效的分析方法。在这里,我们在英国和爱沙尼亚生物库中应用了我们最近开发的贝叶斯分组混合回归模型(GMRM),并在 21 个可遗传性状中获得了迄今为止报道的最高基因组预测准确性。与其他方法相比,GMRM 的准确性比在 LDAK 或 LDPred-funct 软件中运行的注释预测模型分别高出 15%(SE 为 7%)和 14%(SE 为 2%),比没有将单核苷酸多态性(SNP)标记分组到次要等位基因频率-连锁不平衡(MAF-LD)注释类别中的基线 BayesR 模型高出 18%(SE 为 3%)。对于身高,在英国生物库的保留样本中的预测准确性为 47%,这是估计值的 76%。然后,我们扩展我们的 GMRM 预测模型,为全基因组关联(GWAS)发现提供混合线性模型关联(MLMA)SNP 标记估计值,这将在无关联的英国生物库个体中检测到的独立基因座增加到 16162 个,而 BoltLMM 和 Regenie 分别为 10550 个和 10095 个,分别增加了 62%和 65%。与基线 BayesR 模型相比,在所有性状中,预测准确性每提高 1%,领先标记的平均[Formula: see text]值就会增加 15.24(SE 为 0.41)。因此,我们表明,针对 SNP 标记的 MAF 和 LD 差异建模遗传关联,并结合基因组功能的先验知识,对于大规模个体水平研究中的基因组预测和发现都很重要。