Laboratory of Genetic Breeding and Molecular Biology, Southwest Forestry University, Kunming, China.
National Wetland Ecosystem Fixed Research Station of Yunnan Dianchi, Southwest Forestry University, Kunming, China.
Front Endocrinol (Lausanne). 2022 Aug 10;13:957742. doi: 10.3389/fendo.2022.957742. eCollection 2022.
N6-methyladenosine (m6A) modification is a critical epigenetic modification in eukaryotes and involves several biological processes and occurrences of diseases. However, the roles and regulatory mechanisms of m6A regulators in osteoporosis (OP) remain unclear. Thus, the purpose of this study is to explore the roles and mechanisms of m6A regulators in OP.
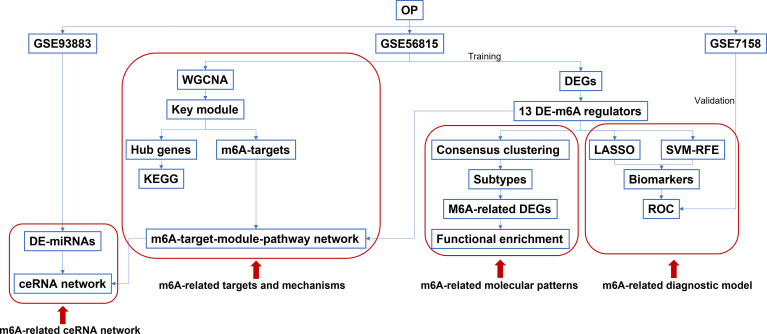
The mRNA and microRNA (miRNA) expression profiles were respectively obtained from GSE56815, GSE7158, and GSE93883 datasets in Gene Expression Omnibus (GEO). The differential expression of 21 m6A regulators between high-bone mineral density (BMD) and low-BMD women was identified. Then, a consensus clustering of low-BMD women was performed based on differentially expressed (DE)-m6A regulators. The m6A-related differentially expressed genes (DEGs), the differentially expressed miRNAs (DE-miRNAs), and biological functions were investigated. Moreover, a weighted gene co-expression network analysis (WGCNA) was constructed to identify the OP-related hub modules, hub genes, and the functional pathways. Then, an m6A regulator-target-pathway network and the competing endogenous RNA (ceRNA) network in key modules were constructed. A least absolute shrinkage and selection operation (LASSO) Cox regression model and a Support Vector Machine-Recursive Feature Elimination (SVM-RFE) model were constructed to identify the candidate genes for OP prediction. The receiver operator characteristic (ROC) curves were used to validate the performances of predictive models and candidate genes.
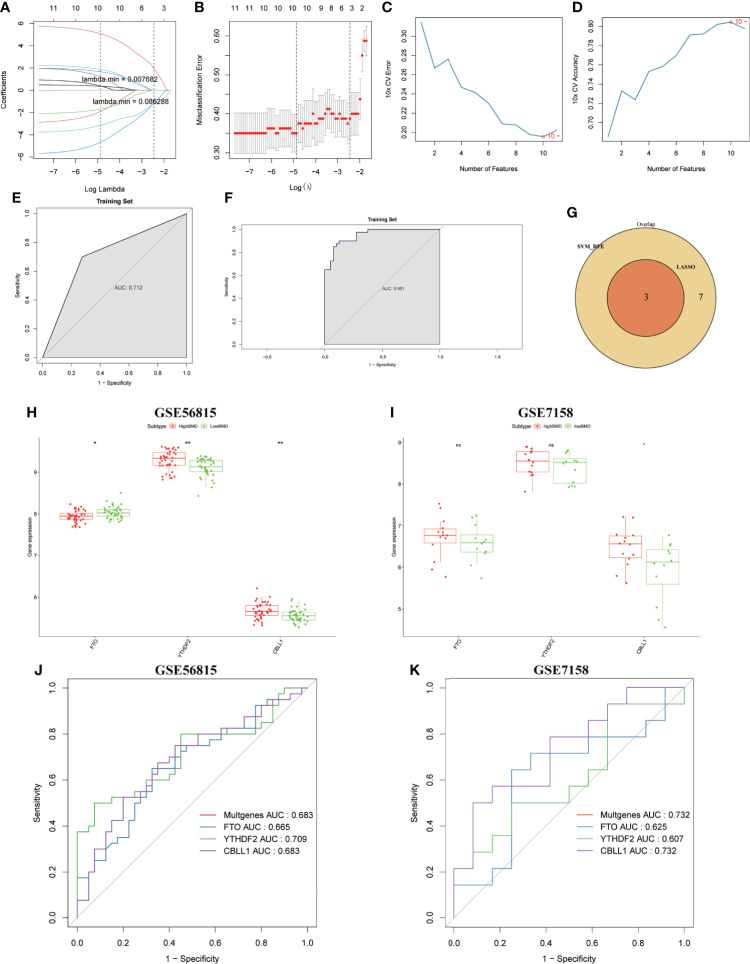
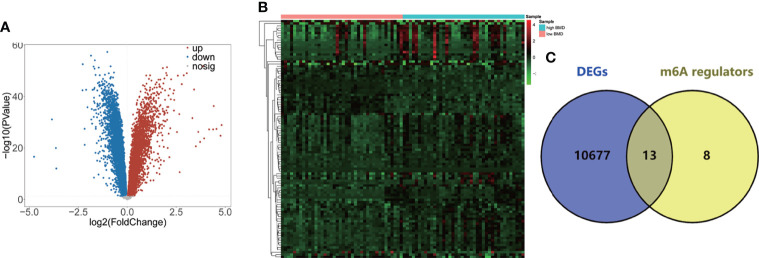
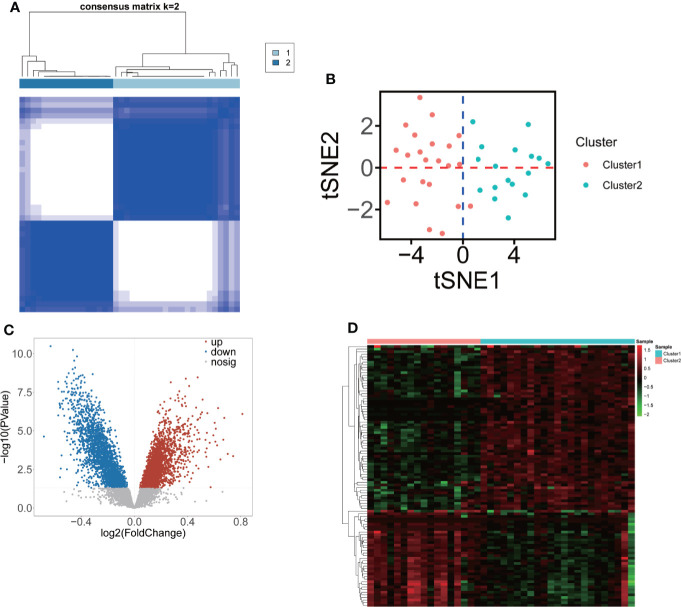
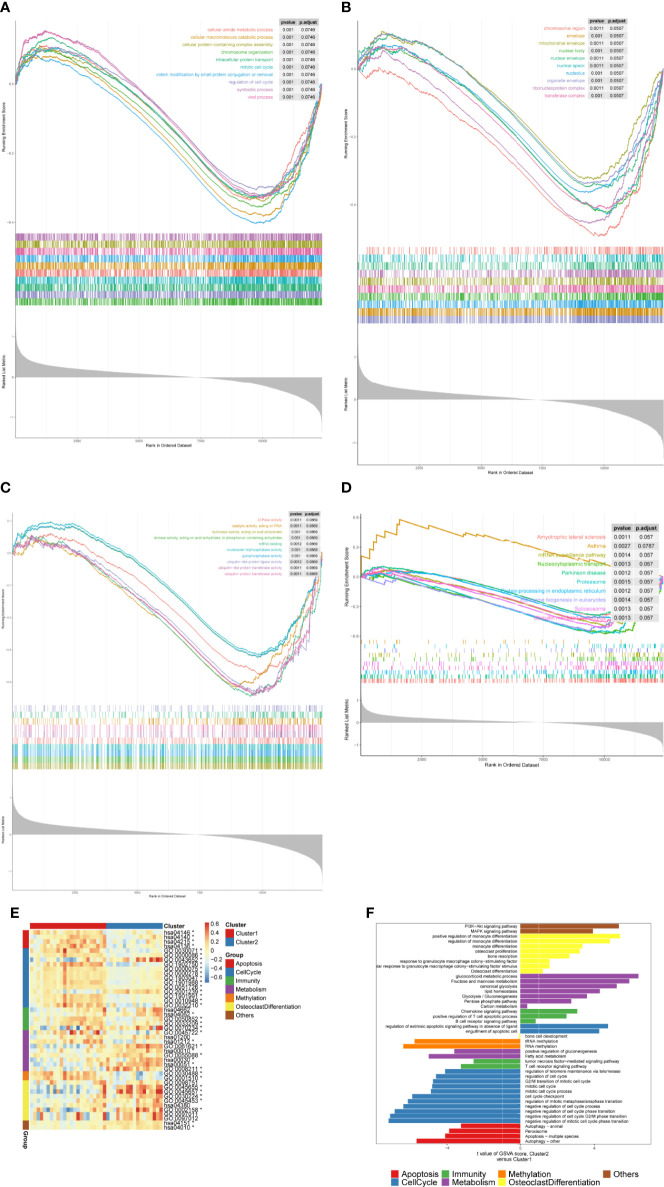
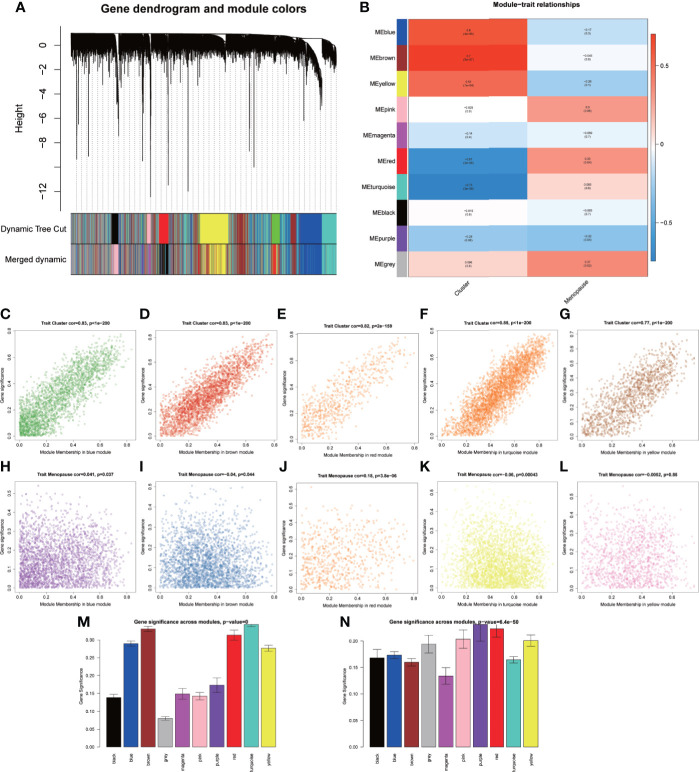
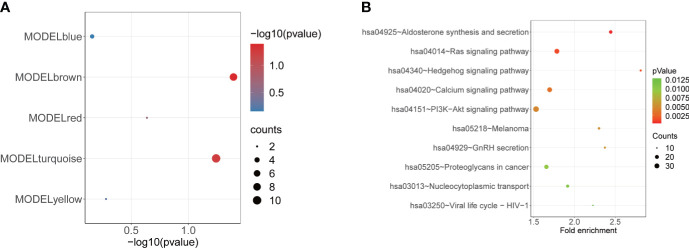
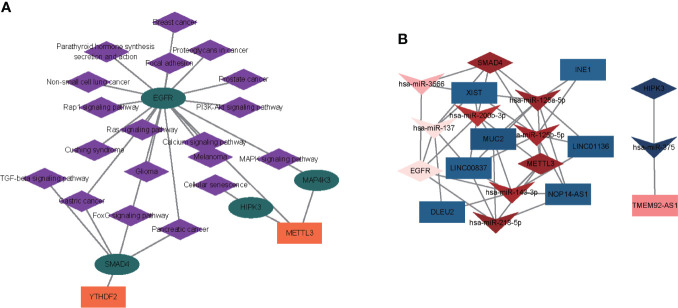
A total of 10,520 DEGs, 13 DE-m6A regulators, and 506 DE-miRNAs between high-BMD and low-BMD women were identified. Two m6A-related subclusters with 13 DE-m6A regulators were classified for OP. There were 5,260 m6A-related DEGs identified between two m6A-related subclusters, the PI3K-Akt, MAPK, and immune-related pathways, and bone metabolism was mainly enriched in cluster 2. Cell cycle-related pathways, RNA methylation, and cell death-related pathways were significantly involved in cluster 1. Five modules were identified as key modules based on WGCNA, and an m6A regulator-target gene-pathway network and the ceRNA network were constructed in module brown. Moreover, three m6A regulators (FTO, YTHDF2, and CBLL1) were selected as the candidate genes for OP.
M6A regulators play an important role in the occurrences and diagnosis of OP.
N6-甲基腺苷(m6A)修饰是真核生物中一种关键的表观遗传修饰,涉及多种生物学过程和疾病的发生。然而,m6A 调节剂在骨质疏松症(OP)中的作用和调节机制尚不清楚。因此,本研究旨在探讨 m6A 调节剂在 OP 中的作用和机制。
从基因表达综合数据库(GEO)中的 GSE56815、GSE7158 和 GSE93883 数据集分别获得 mRNA 和 microRNA(miRNA)表达谱。鉴定出高骨密度(BMD)和低-BMD 女性之间 21 种 m6A 调节剂的差异表达。然后,基于差异表达(DE)-m6A 调节剂对低-BMD 女性进行共识聚类。研究 m6A 相关差异表达基因(DEGs)、差异表达 miRNA(DE-miRNAs)和生物学功能。此外,构建加权基因共表达网络分析(WGCNA)以鉴定与 OP 相关的枢纽模块、枢纽基因和功能途径。然后,构建关键模块中的 m6A 调节剂-靶途径网络和竞争内源 RNA(ceRNA)网络。构建最小绝对收缩和选择操作(LASSO)Cox 回归模型和支持向量机-递归特征消除(SVM-RFE)模型,以鉴定 OP 预测的候选基因。使用接收器操作特征(ROC)曲线验证预测模型和候选基因的性能。
共鉴定出高-BMD 和低-BMD 女性之间的 10520 个 DEGs、13 个 DE-m6A 调节剂和 506 个 DE-miRNAs。两个 m6A 相关亚群被分为 OP。在两个 m6A 相关亚群之间鉴定出 5260 个 m6A 相关 DEG,涉及 PI3K-Akt、MAPK 和免疫相关途径,骨代谢主要在簇 2 中富集。细胞周期相关途径、RNA 甲基化和细胞死亡相关途径在簇 1 中显著参与。基于 WGCNA 鉴定出 5 个关键模块,并构建了模块棕色的 m6A 调节剂-靶基因-途径网络和 ceRNA 网络。此外,选择了三个 m6A 调节剂(FTO、YTHDF2 和 CBLL1)作为 OP 的候选基因。
m6A 调节剂在 OP 的发生和诊断中起重要作用。