Guangdong Provincial Key Lab of Agro-animal Genomics and Molecular Breeding, National Engineering Research Center for Breeding Swine Industry, College of Animal Science, South China Agricultural University, 510642, Guangzhou, China.
Department of Animal Science, College of Animal Science, Zhejiang University, 310058, Hangzhou, China.
BMC Genomics. 2022 Nov 30;23(1):786. doi: 10.1186/s12864-022-09036-z.
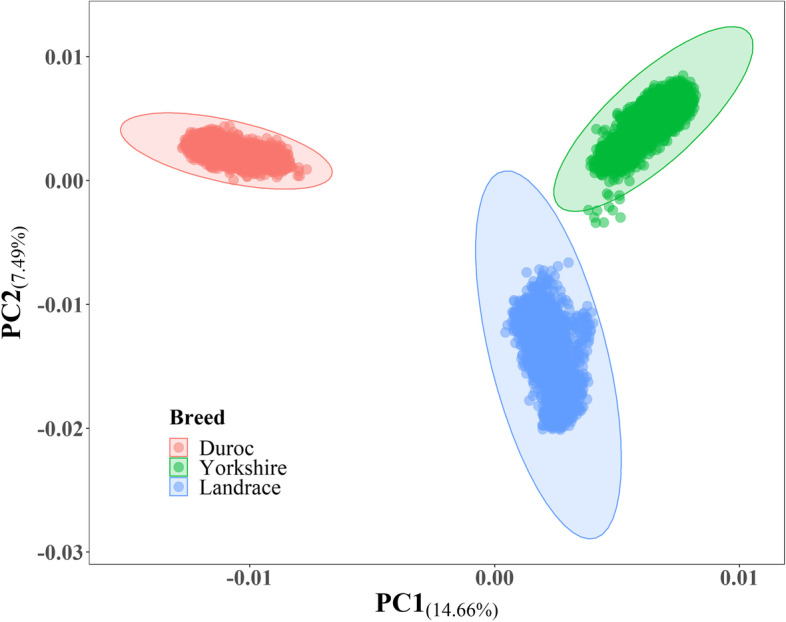
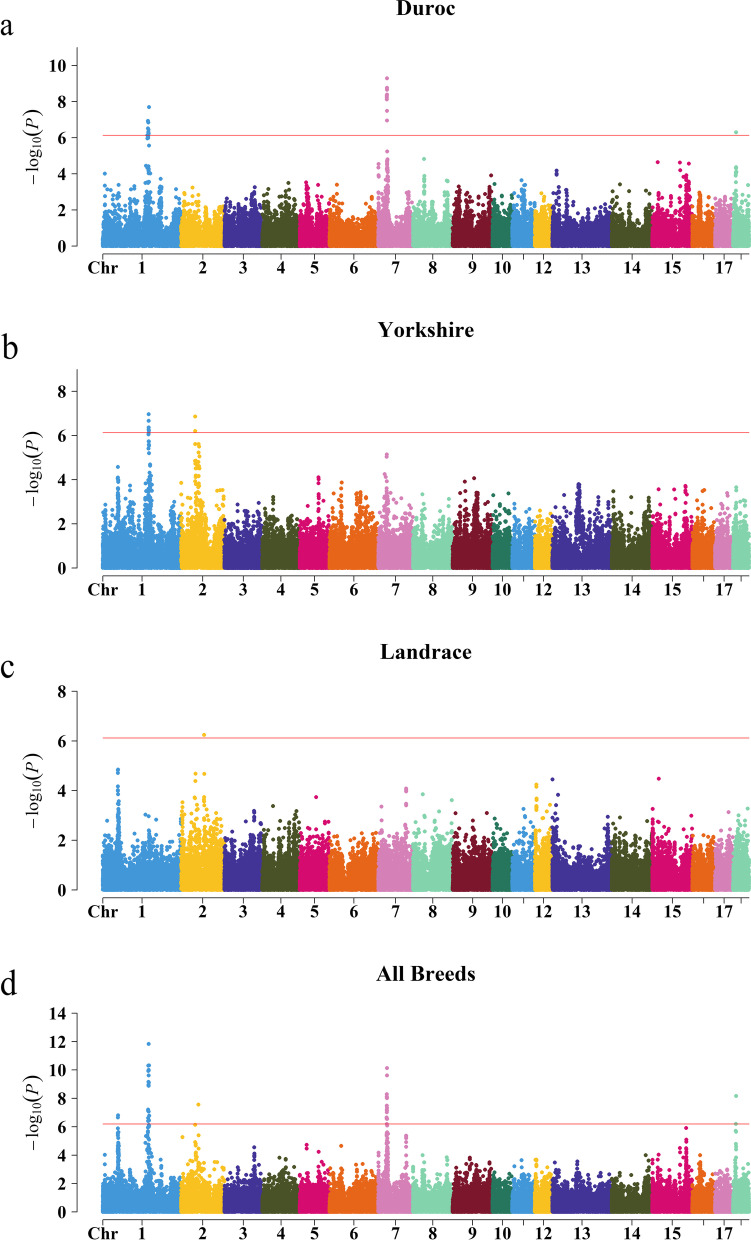
Average backfat thickness (BFT) is a critical complex trait in pig and an important indicator for fat deposition and lean rate. Usually, genome-wide association study (GWAS) was used to discover quantitative trait loci (QTLs) of BFT in a single population. However, the power of GWAS is limited by sample size in a single population. Alternatively, meta-analysis of GWAS (metaGWAS) is an attractive method to increase the statistical power by integrating data from multiple breeds and populations. The aim of this study is to identify shared genetic characterization of BFT across breeds in pigs via metaGWAS. RESULTS: In this study, we performed metaGWAS on BFT using 15,353 pigs (5,143 Duroc, 7,275 Yorkshire, and 2,935 Landrace) from 19 populations. We detected 40 genome-wide significant SNPs (Bonferroni corrected P < 0.05) and defined five breed-shared QTLs in across-breed metaGWAS. Markers within the five QTL regions explained 7 ~ 9% additive genetic variance and showed strong heritability enrichment. Furthermore, by integrating information from multiple bioinformatics databases, we annotated 46 candidate genes located in the five QTLs. Among them, three important (MC4R, PPARD, and SLC27A1) and seven suggestive candidate genes (PHLPP1, NUDT3, ILRUN, RELCH, KCNQ5, ITPR3, and U3) were identified.
QTLs and candidate genes underlying BFT across breeds were identified via metaGWAS from multiple populations. Our findings contribute to the understanding of the genetic architecture of BFT and the regulating mechanism underlying fat deposition in pigs.
平均背膘厚(BFT)是猪的一个关键复杂性状,是脂肪沉积和瘦肉率的重要指标。通常,全基因组关联研究(GWAS)用于在单个群体中发现 BFT 的数量性状位点(QTL)。然而,GWAS 的功效受到单个群体中样本量的限制。相反,GWAS 的荟萃分析(metaGWAS)是一种通过整合来自多个品种和群体的数据来增加统计功效的有吸引力的方法。本研究的目的是通过 metaGWAS 鉴定猪在不同品种间 BFT 的共享遗传特征。
在本研究中,我们使用来自 19 个群体的 15353 头猪(5143 头杜洛克、7275 头约克夏和 2935 头长白)进行了 BFT 的 metaGWAS。我们检测到 40 个全基因组显著 SNP(Bonferroni 校正后 P < 0.05),并在跨品种 metaGWAS 中定义了 5 个品种共享的 QTL。五个 QTL 区域内的标记解释了 7%~9%的加性遗传方差,并显示出强烈的遗传力富集。此外,通过整合来自多个生物信息学数据库的信息,我们注释了位于五个 QTL 中的 46 个候选基因。其中,三个重要(MC4R、PPARD 和 SLC27A1)和七个提示候选基因(PHLPP1、NUDT3、ILRUN、RELCH、KCNQ5、ITPR3 和 U3)被鉴定出来。
通过来自多个群体的 metaGWAS 鉴定了跨品种 BFT 的 QTL 和候选基因。我们的研究结果有助于理解 BFT 的遗传结构以及猪脂肪沉积的调控机制。