Sundin Iiris, Voronov Alexey, Xiao Haoping, Papadopoulos Kostas, Bjerrum Esben Jannik, Heinonen Markus, Patronov Atanas, Kaski Samuel, Engkvist Ola
Department of Computer Science, Aalto University, Espoo, Finland.
Molecular AI, Discovery Sciences, R&D, AstraZeneca, Gothenburg, Sweden.
J Cheminform. 2022 Dec 28;14(1):86. doi: 10.1186/s13321-022-00667-8.
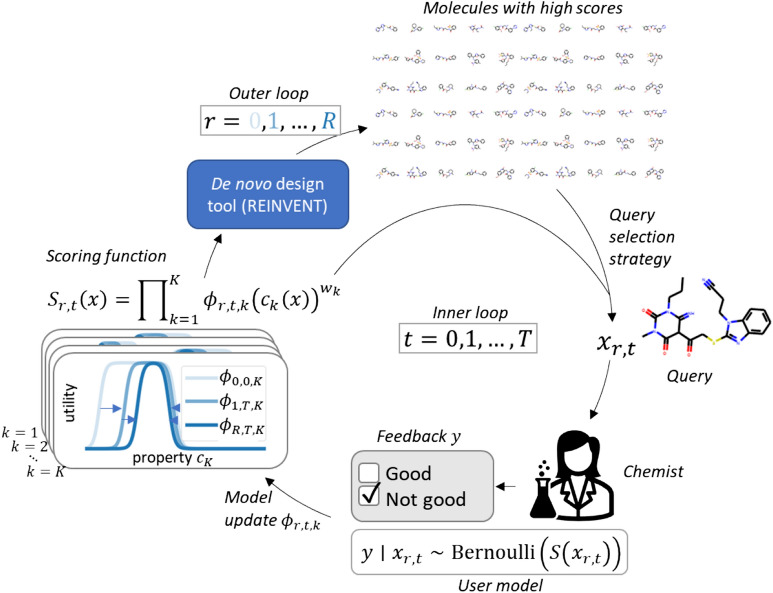
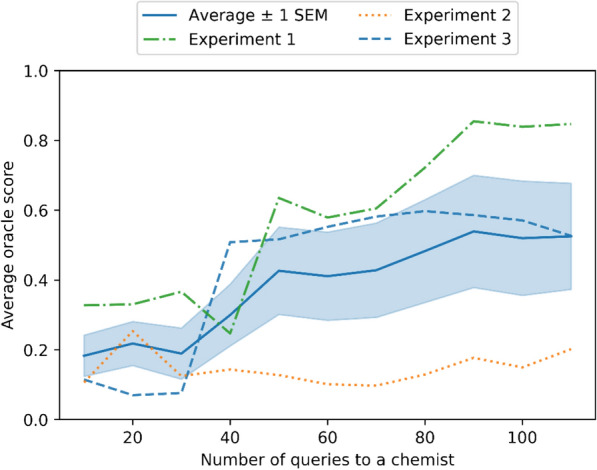
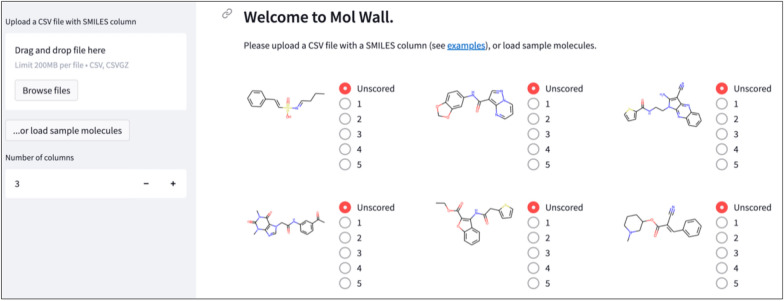
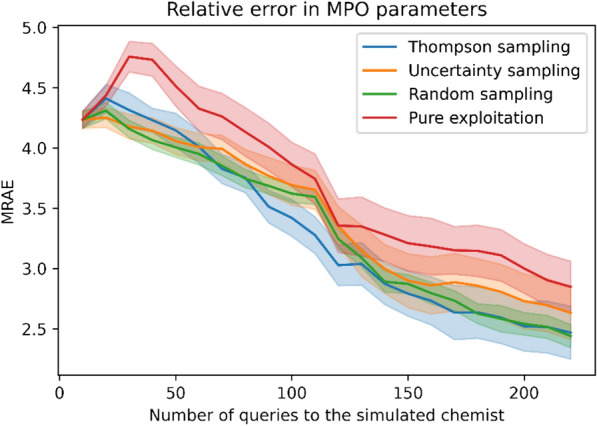
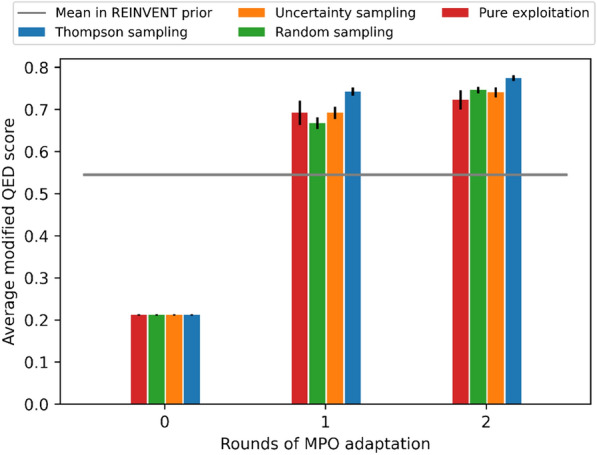
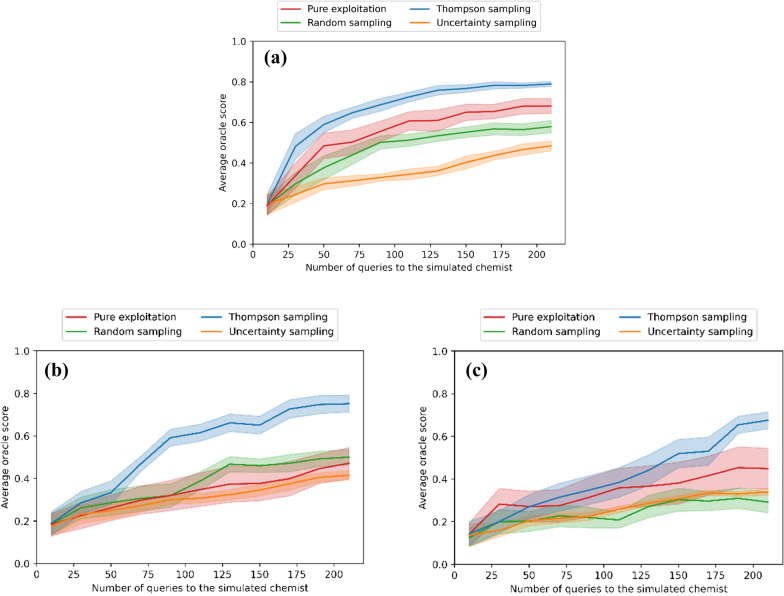
A de novo molecular design workflow can be used together with technologies such as reinforcement learning to navigate the chemical space. A bottleneck in the workflow that remains to be solved is how to integrate human feedback in the exploration of the chemical space to optimize molecules. A human drug designer still needs to design the goal, expressed as a scoring function for the molecules that captures the designer's implicit knowledge about the optimization task. Little support for this task exists and, consequently, a chemist usually resorts to iteratively building the objective function of multi-parameter optimization (MPO) in de novo design. We propose a principled approach to use human-in-the-loop machine learning to help the chemist to adapt the MPO scoring function to better match their goal. An advantage is that the method can learn the scoring function directly from the user's feedback while they browse the output of the molecule generator, instead of the current manual tuning of the scoring function with trial and error. The proposed method uses a probabilistic model that captures the user's idea and uncertainty about the scoring function, and it uses active learning to interact with the user. We present two case studies for this: In the first use-case, the parameters of an MPO are learned, and in the second use-case a non-parametric component of the scoring function to capture human domain knowledge is developed. The results show the effectiveness of the methods in two simulated example cases with an oracle, achieving significant improvement in less than 200 feedback queries, for the goals of a high QED score and identifying potent molecules for the DRD2 receptor, respectively. We further demonstrate the performance gains with a medicinal chemist interacting with the system.
从头开始的分子设计工作流程可以与强化学习等技术一起使用,以探索化学空间。该工作流程中仍有待解决的一个瓶颈是如何在化学空间探索中整合人类反馈以优化分子。人类药物设计师仍需要设计目标,将其表示为分子的评分函数,该函数捕获设计师关于优化任务的隐性知识。目前对此任务的支持很少,因此,化学家通常在从头设计中采用迭代构建多参数优化(MPO)目标函数的方法。我们提出了一种有原则的方法,使用人在回路机器学习来帮助化学家调整MPO评分函数,以更好地匹配他们的目标。一个优点是,该方法可以在用户浏览分子生成器的输出时直接从用户反馈中学习评分函数,而不是目前通过反复试验对手动调整评分函数。所提出的方法使用一个概率模型来捕捉用户对评分函数的想法和不确定性,并使用主动学习与用户进行交互。我们为此展示了两个案例研究:在第一个用例中,学习MPO的参数,在第二个用例中,开发评分函数的非参数组件以捕捉人类领域知识。结果表明,在两个带有预言机的模拟示例案例中,这些方法是有效的,分别针对高QED分数和识别DRD2受体的强效分子的目标,在不到200次反馈查询中取得了显著改进。我们进一步展示了药物化学家与系统交互时的性能提升。