Manrique Pedro D, Chakraborty Srirupa, Henderson Rory, Edwards Robert J, Mansbach Rachael, Nguyen Kien, Stalls Victoria, Saunders Carrie, Mansouri Katayoun, Acharya Priyamvada, Korber Bette, Gnanakaran S
Theoretical Biology and Biophysics Group, Los Alamos National Laboratory, Los Alamos, NM 87545, USA.
Center for Nonlinear Studies, Los Alamos National Laboratory, Los Alamos, NM 87545, USA.
iScience. 2023 Jan 20;26(1):105855. doi: 10.1016/j.isci.2022.105855. Epub 2022 Dec 26.
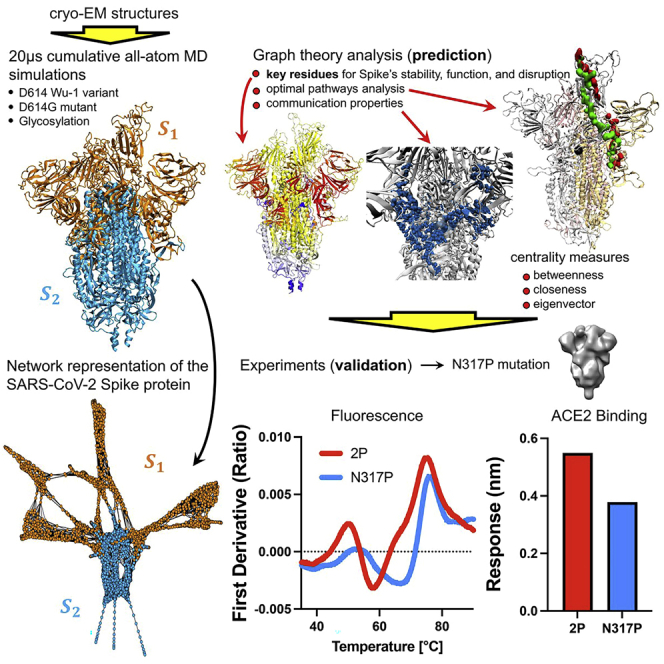
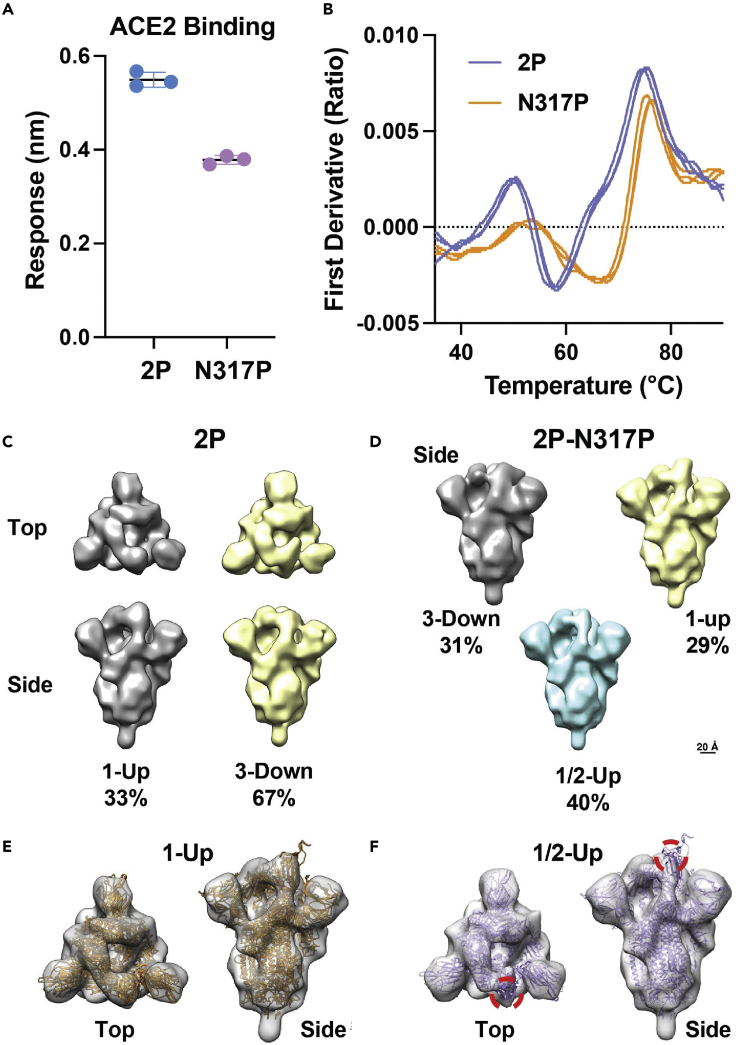
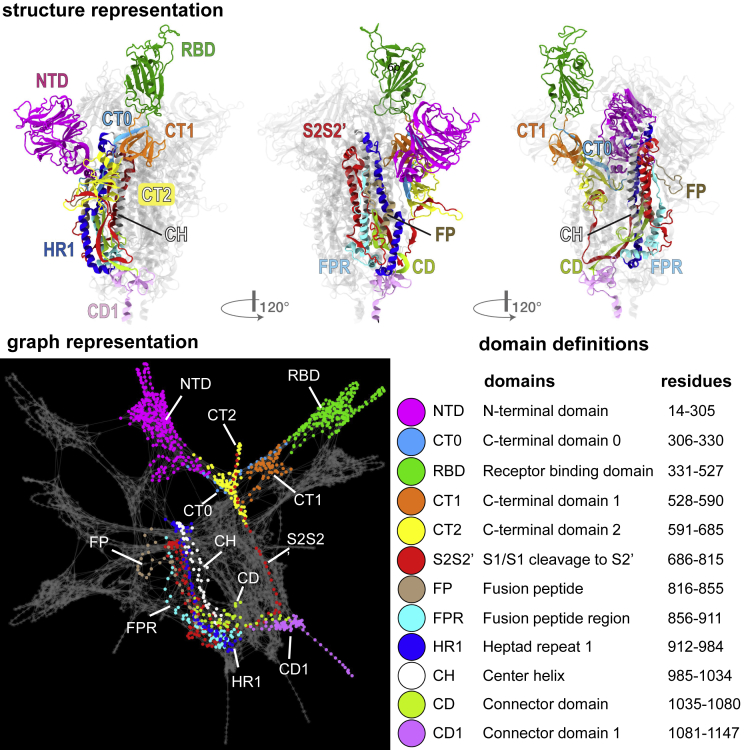
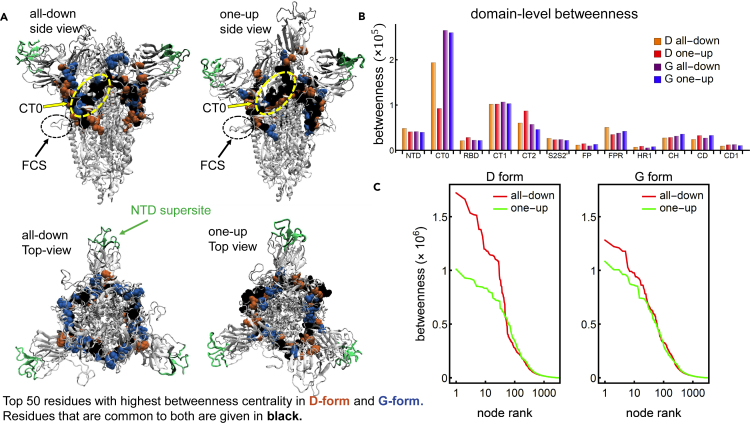
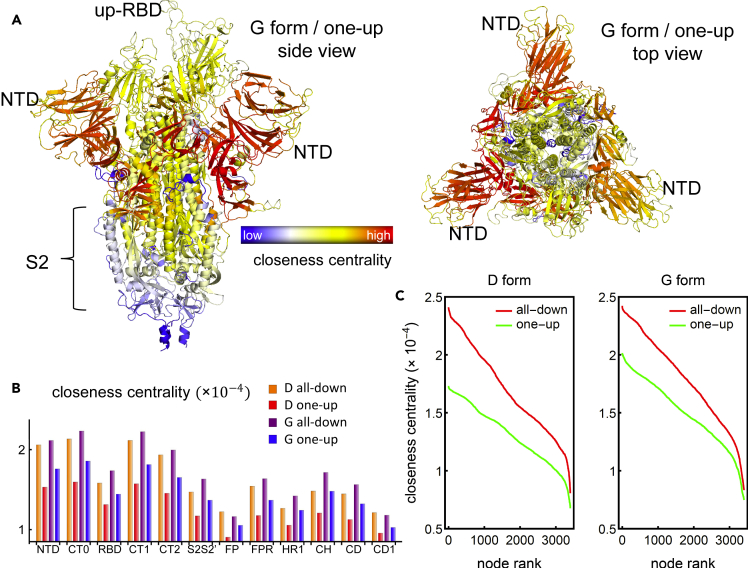
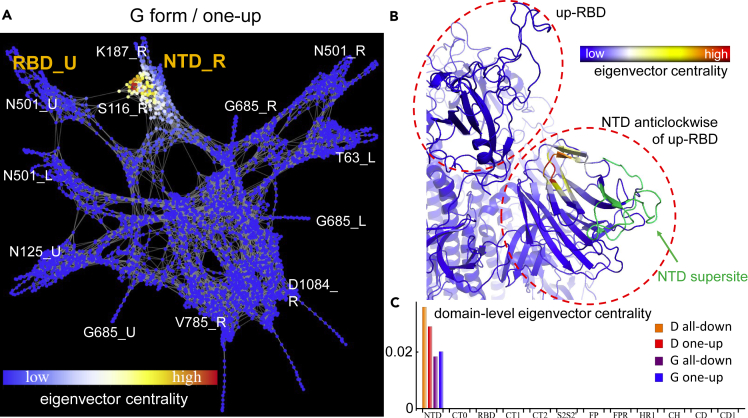
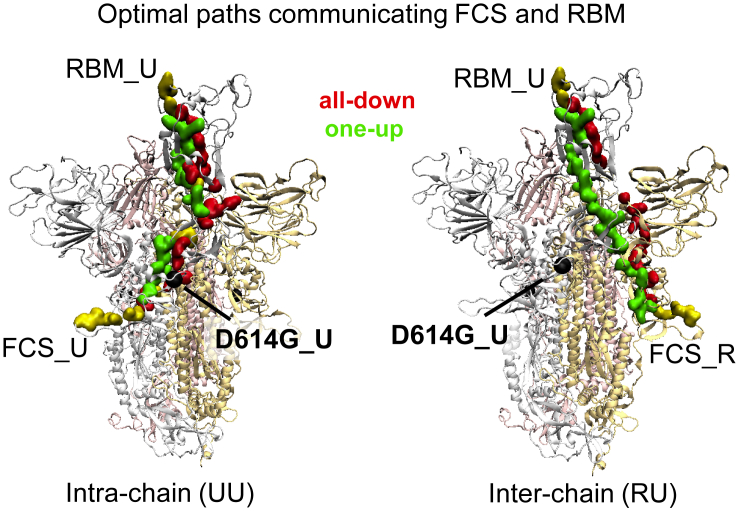
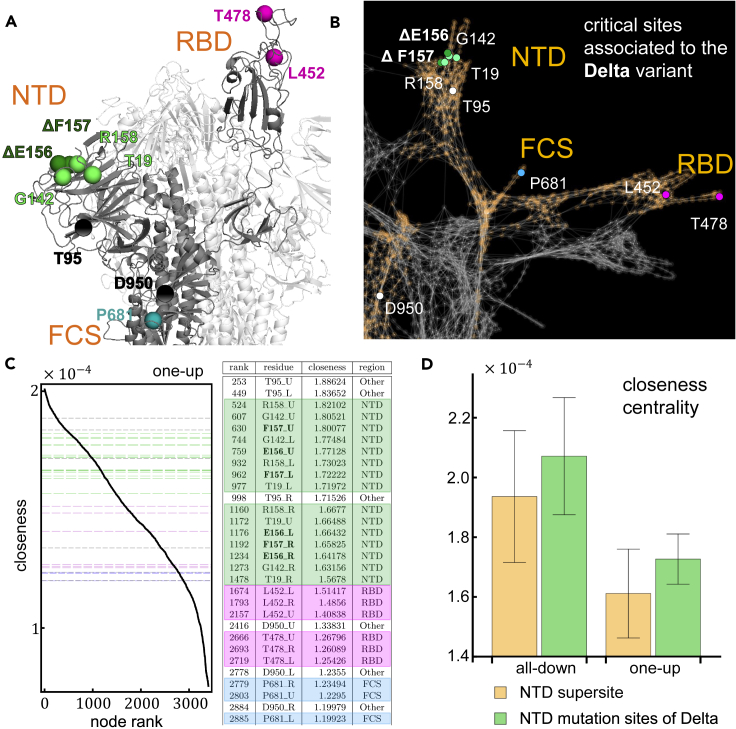
The COVID-19 pandemic, caused by the SARS-CoV-2 virus, has triggered myriad efforts to understand the structure and dynamics of this complex pathogen. The spike glycoprotein of SARS-CoV-2 is a significant target for immunogens as it is the means by which the virus enters human cells, while simultaneously sporting mutations responsible for immune escape. These functional and escape processes are regulated by complex molecular-level interactions. Our study presents quantitative insights on domain and residue contributions to allosteric communication, immune evasion, and local- and global-level control of functions through the derivation of a weighted graph representation from all-atom MD simulations. Focusing on the ancestral form and the D614G-variant, we provide evidence of the utility of our approach by guiding the selection of a mutation that alters the spike's stability. Taken together, the network approach serves as a valuable tool to evaluate communication "hot-spots" in proteins to guide design of stable immunogens.
由严重急性呼吸综合征冠状病毒2(SARS-CoV-2)引起的2019冠状病毒病(COVID-19)大流行引发了无数旨在了解这种复杂病原体的结构和动态的研究。SARS-CoV-2的刺突糖蛋白是免疫原的重要靶点,因为它是病毒进入人体细胞的途径,同时还带有负责免疫逃逸的突变。这些功能和逃逸过程受复杂的分子水平相互作用调控。我们的研究通过从全原子分子动力学模拟中推导加权图表示,对结构域和残基在变构通讯、免疫逃逸以及功能的局部和全局控制方面的贡献提供了定量见解。聚焦于原始形式和D614G变体,我们通过指导选择改变刺突稳定性的突变,证明了我们方法的实用性。综合来看,网络方法是评估蛋白质中通讯“热点”以指导稳定免疫原设计的宝贵工具。