Gong Linjing, Shen Yue, Wang Sijiao, Wang Xinyuan, Ji Haiying, Wu Xu, Hu Lijuan, Zhu Lei
Department of Respiratory and Critical Care Medicine, West China Hospital, Sichuan University, No 37 Guoxue Alley, 610041, Chengdu, Sichuan, China.
Department of Pulmonary Medicine, Zhongshan Hospital, Fudan University, 200032, Shanghai, China.
Cell Death Discov. 2023 Jan 18;9(1):12. doi: 10.1038/s41420-023-01320-5.
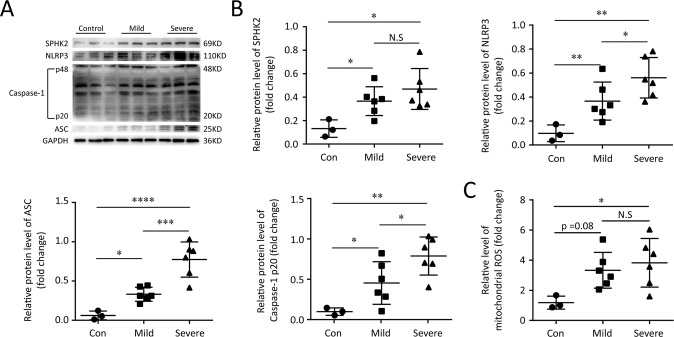
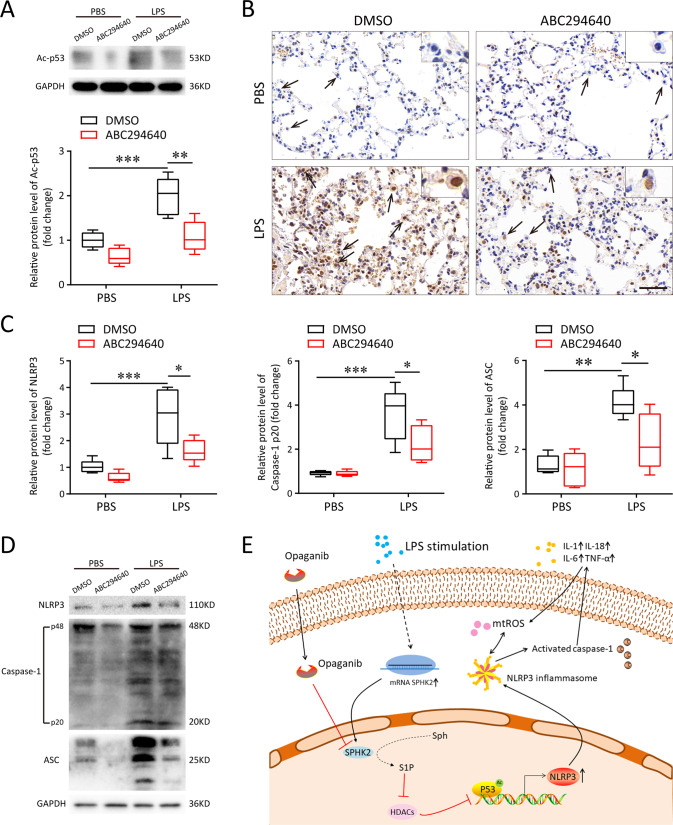
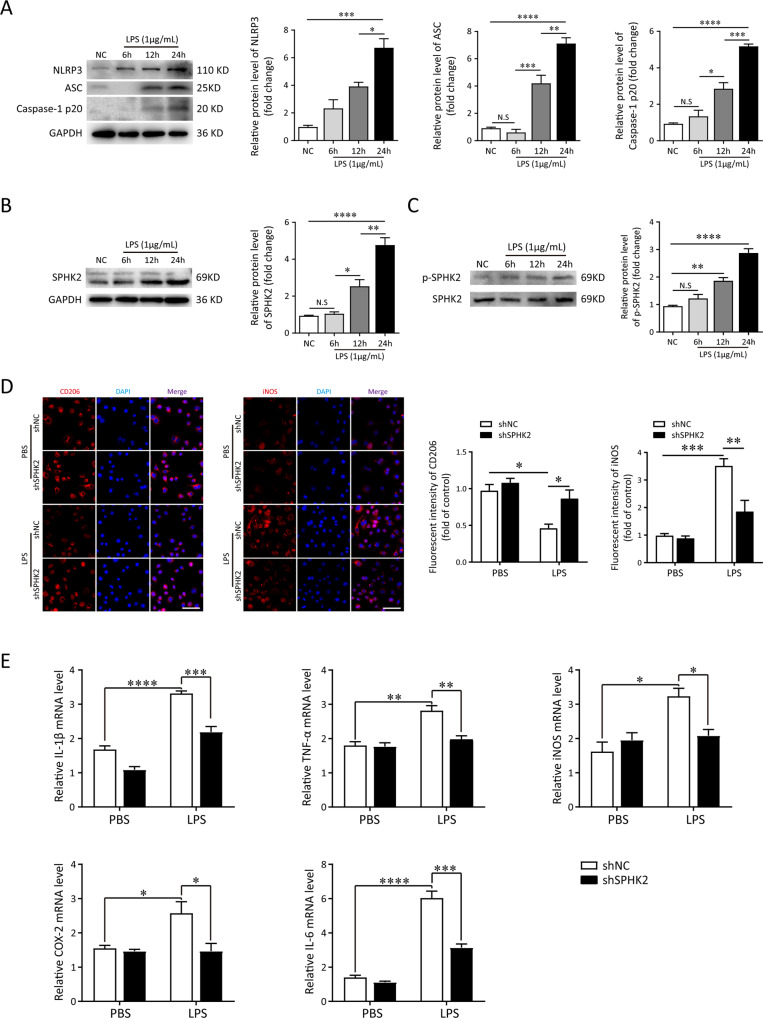
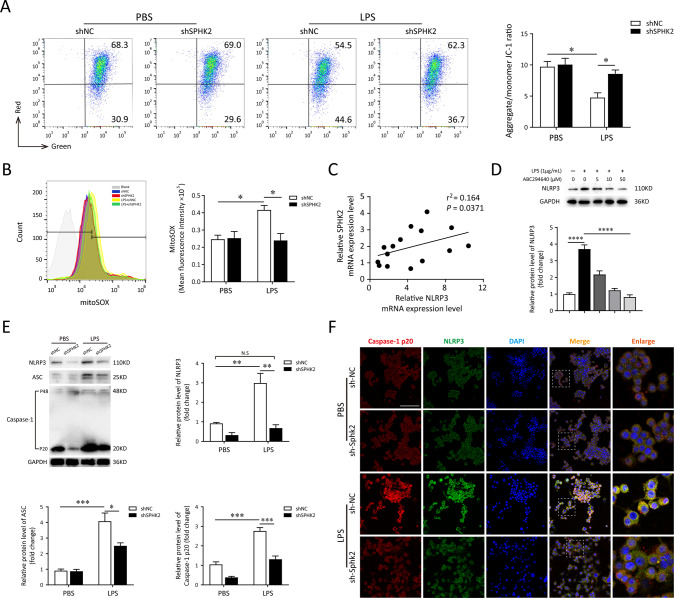
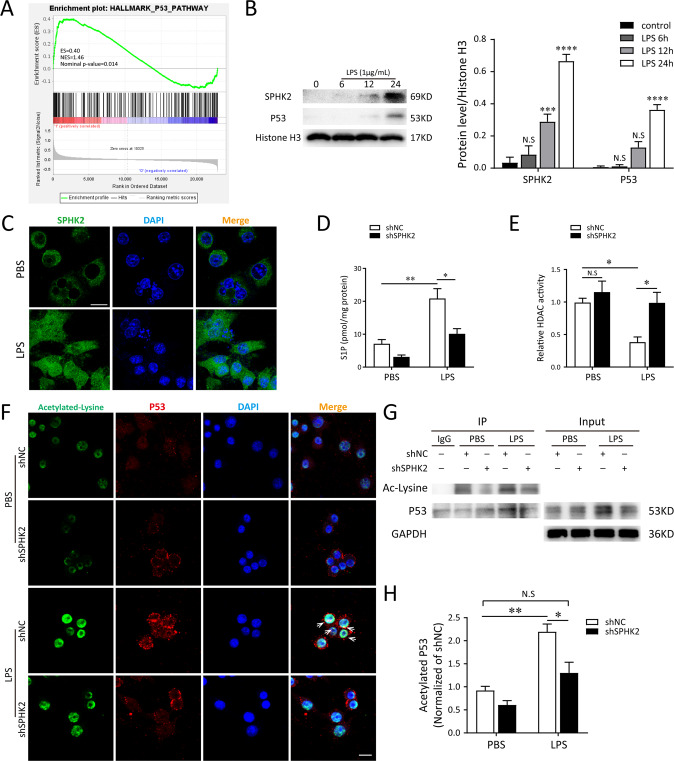
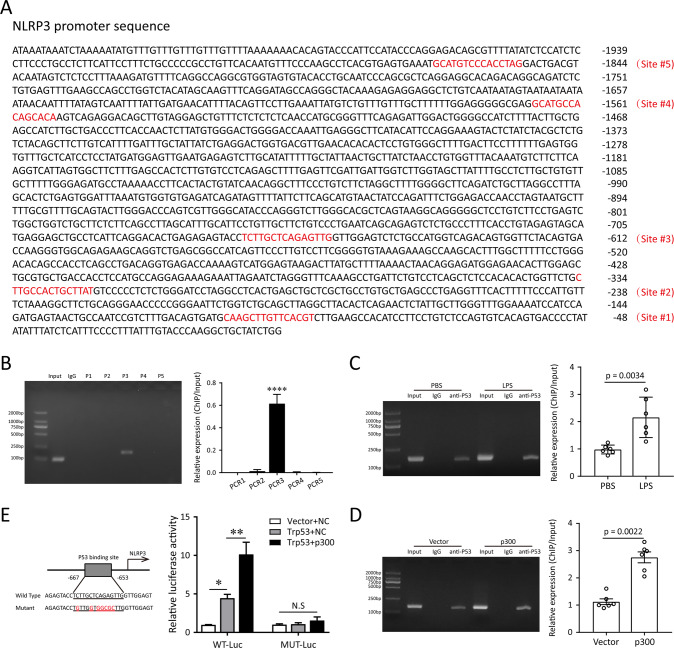
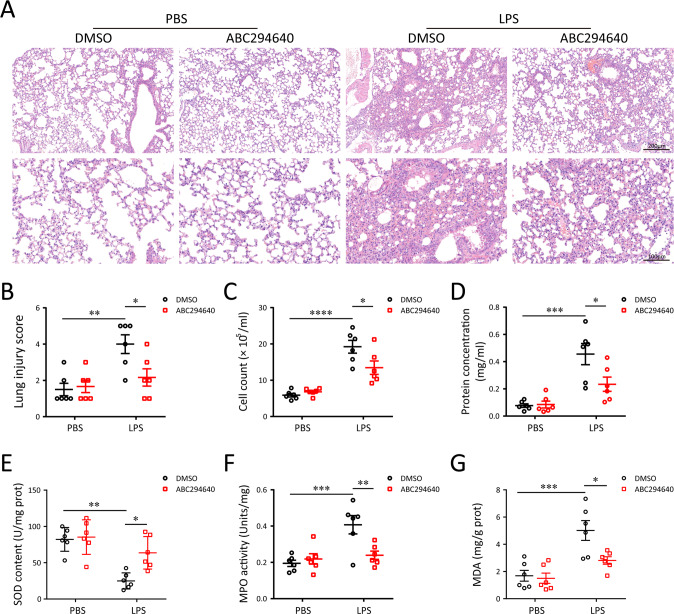
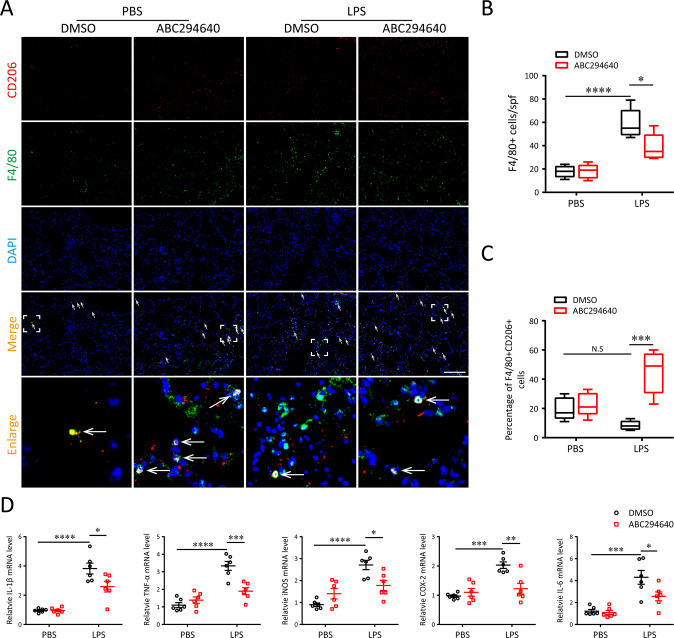
A bulk of evidence identified that macrophages, including resident alveolar macrophages and recruited macrophages from the blood, played an important role in the pathogenesis of acute respiratory distress syndrome (ARDS). However, the molecular mechanisms of macrophages-induced acute lung injury (ALI) by facilitating oxidative stress and inflammatory responses remain unclear. Herein, we noticed that the levels of mitochondrial reactive oxygen species (mtROS), SPHK2 and activated NLRP3 inflammasome were higher in peripheral blood mononuclear cells (PBMCs) of ARDS patients than that in healthy volunteers. Similar observations were recapitulated in LPS-treated RAW264.7 and THP-1 cells. After exposure to LPS, the SPHK2 enzymatic activity, NLRP3 inflammasome activation and mtROS were significantly upregulated in macrophages. Moreover, knockdown SPHK2 via shRNA or inhibition SPHK2 could prominently decrease LPS-induced M1 macrophage polarization, oxidative stress and NLRP3 inflammasome activation. Further study indicated that upregulated SPHK2 could increase nuclear sphingosine-1-phosphate (S1P) levels and then restrict the enzyme activity of HDACs to facilitate p53 acetylation. Acetylation of p53 reinforced its binding to the specific region of the NLRP3 promoter and drove expression of NLRP3. In the in vivo experiments, it was also observed that treating with Opaganib (ABC294640), a specific SPHK2 inhibitor, could observably alleviate LPS-induced ALI, evidencing by lowered infiltration of inflammatory cells, increased M2 macrophages polarization and reduced oxidative damage in lung tissues. Besides, SPHK2 inhibition can also decrease the accumulation of acetylated p53 protein and the activation of NLRP3 inflammasome. Taken together, our results demonstrated for the first time that nuclear S1P can regulate the acetylation levels of non-histone protein through affecting HDACs enzyme activities, linking them to oxidative stress and inflammation in response to environmental signals. These data provide a theoretical basis that SPHK2 may be an effective therapeutic target of ARDS.
大量证据表明,巨噬细胞,包括驻留的肺泡巨噬细胞和从血液中募集的巨噬细胞,在急性呼吸窘迫综合征(ARDS)的发病机制中起重要作用。然而,巨噬细胞通过促进氧化应激和炎症反应诱导急性肺损伤(ALI)的分子机制仍不清楚。在此,我们注意到ARDS患者外周血单核细胞(PBMC)中线粒体活性氧(mtROS)、SPHK2和活化的NLRP3炎性小体水平高于健康志愿者。在LPS处理的RAW264.7和THP-1细胞中也得到了类似的观察结果。暴露于LPS后,巨噬细胞中SPHK2酶活性、NLRP3炎性小体活化和mtROS显著上调。此外,通过shRNA敲低SPHK2或抑制SPHK2可显著降低LPS诱导的M1巨噬细胞极化、氧化应激和NLRP3炎性小体活化。进一步研究表明,上调的SPHK2可增加细胞核鞘氨醇-1-磷酸(S1P)水平,进而限制HDACs的酶活性,促进p53乙酰化。p53的乙酰化增强了其与NLRP3启动子特定区域的结合,并驱动NLRP3的表达。在体内实验中,还观察到用特异性SPHK2抑制剂Opaganib(ABC294640)治疗可明显减轻LPS诱导的ALI,表现为肺组织中炎性细胞浸润减少、M2巨噬细胞极化增加和氧化损伤减轻。此外,抑制SPHK2还可降低乙酰化p53蛋白的积累和NLRP3炎性小体的活化。综上所述,我们的结果首次证明细胞核S1P可通过影响HDACs酶活性来调节非组蛋白的乙酰化水平,将它们与响应环境信号的氧化应激和炎症联系起来。这些数据为SPHK2可能是ARDS的有效治疗靶点提供了理论依据。