Department of Pharmacology (State-Province Key Laboratories of Biomedicine-Pharmaceutics of China, Key Laboratory of Cardiovascular Research, Ministry of Education), College of Pharmacy, Harbin Medical University, Heilongjiang, China (Y.Y., Y.Z., J.Y., M.Z., T.T., Y.J., X.L., G.X., X.L., X.Z., S.L., X.H., Z.L., Y.G., L.Z., H.B., Z. Zhou, J.S., G.Y., L.X., H.S., Y.L., B.Y., Z.P.).
Department of Cardiology, Xiamen Key Laboratory of Cardiac Electrophysiology, Xiamen Institute of Cardiovascular Diseases, The First Affiliated Hospital of Xiamen University, School of Medicine, Xiamen University, China (Y.Y.).
Circ Res. 2023 Jan 20;132(2):208-222. doi: 10.1161/CIRCRESAHA.122.321153. Epub 2022 Dec 30.
ASPP1 (apoptosis stimulating of p53 protein 1) is critical in regulating cell apoptosis as a cofactor of p53 to promote its transcriptional activity in the nucleus. However, whether cytoplasmic ASPP1 affects p53 nuclear trafficking and its role in cardiac diseases remains unknown. This study aims to explore the mechanism by which ASPP1 modulates p53 nuclear trafficking and the subsequent contribution to cardiac ischemia/reperfusion (I/R) injury.
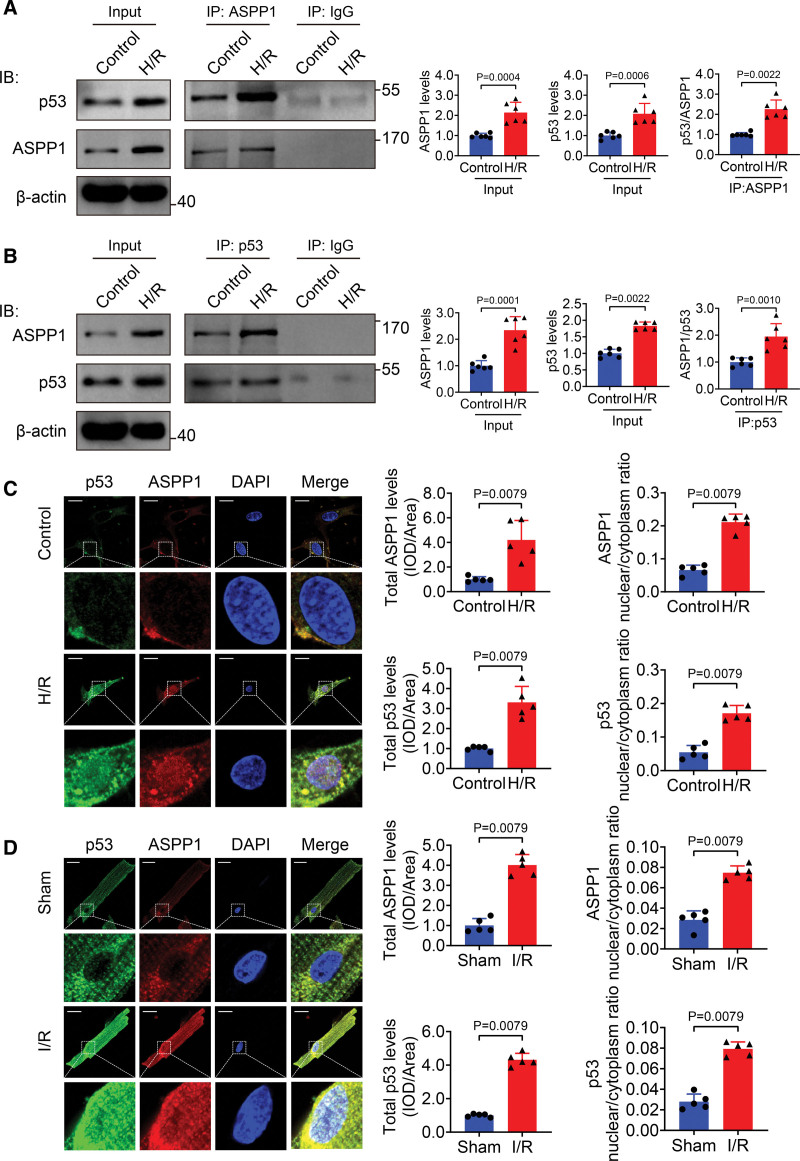
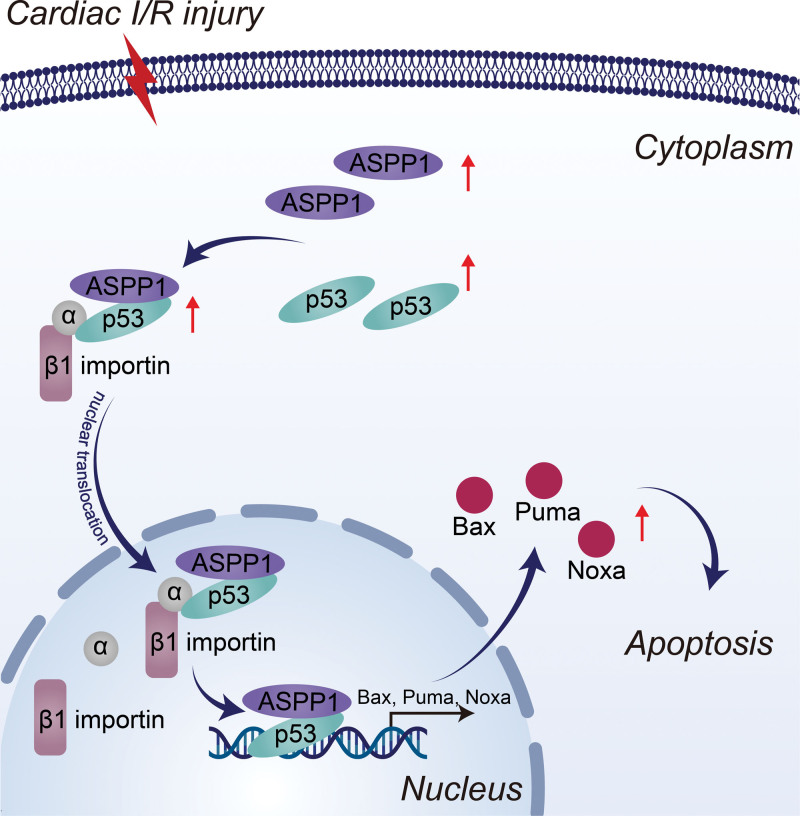
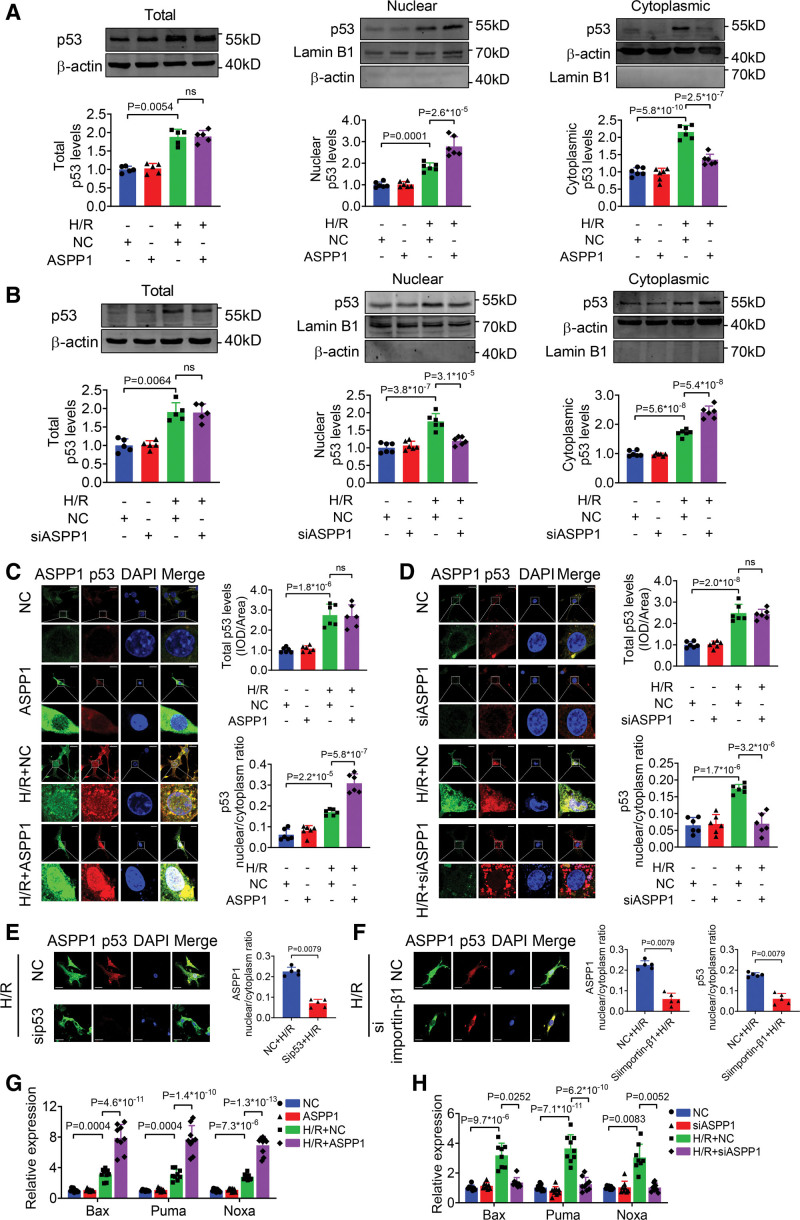
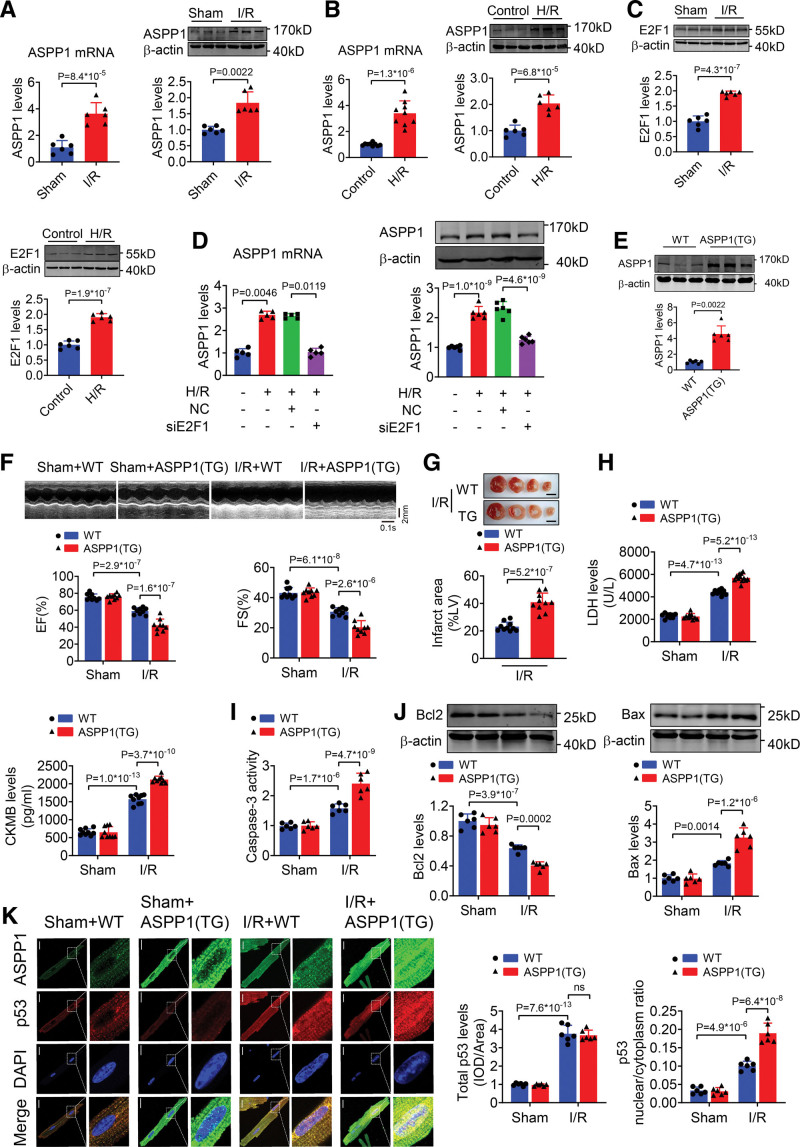
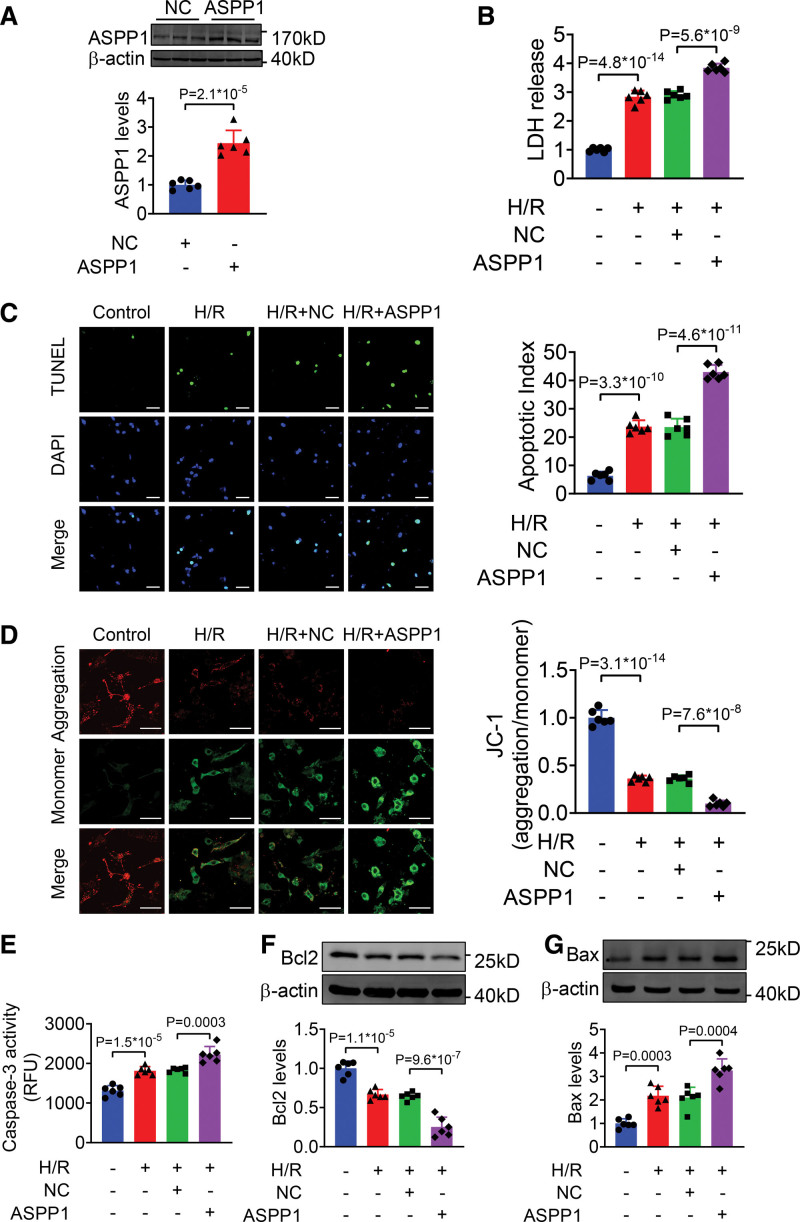
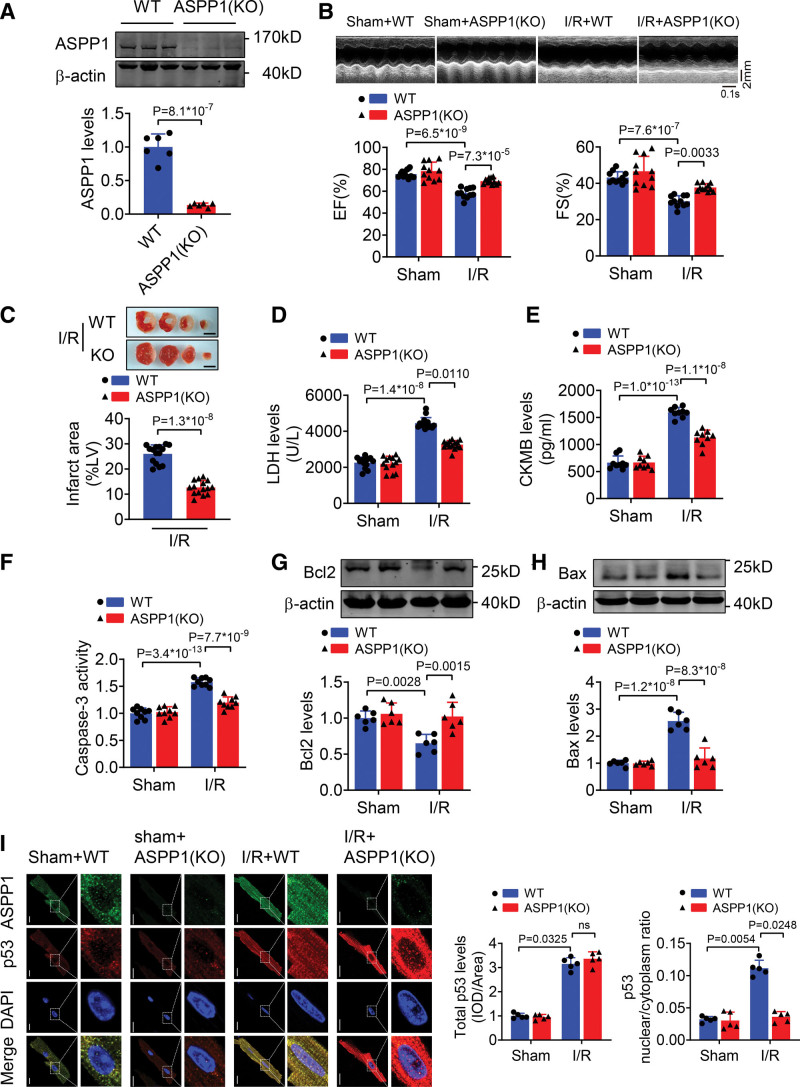
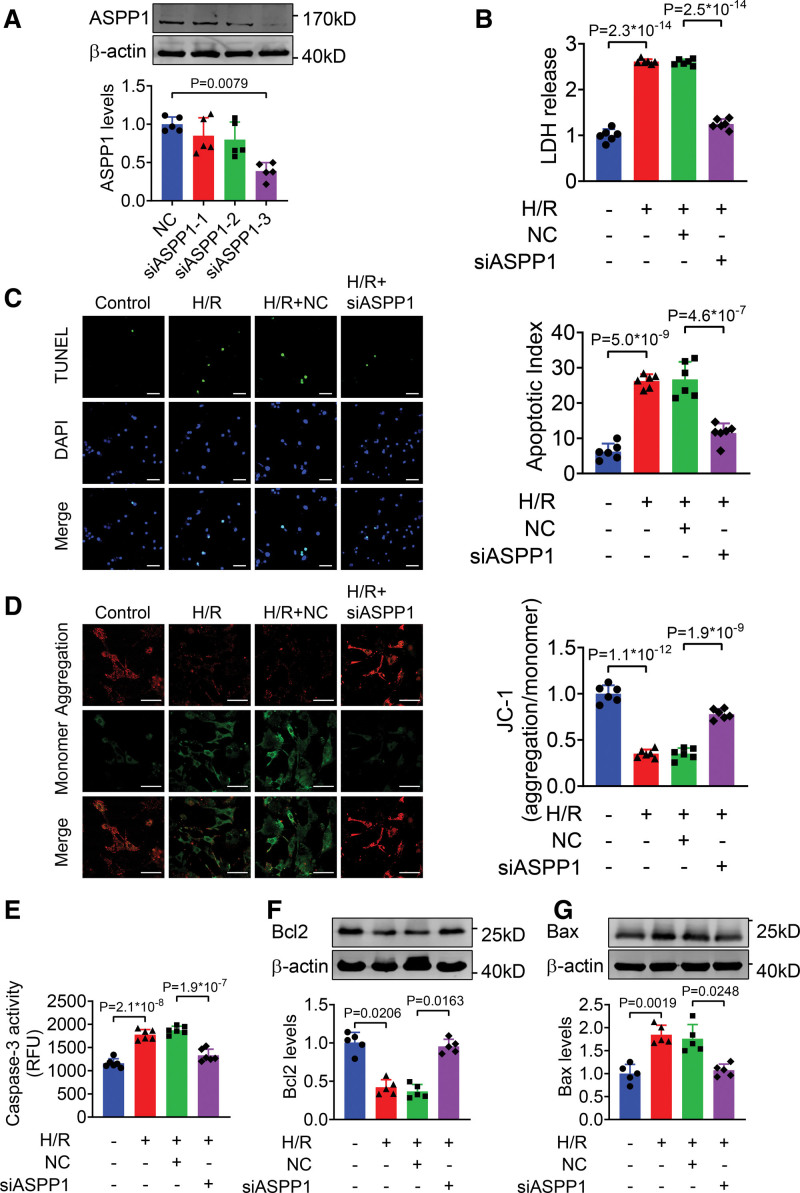
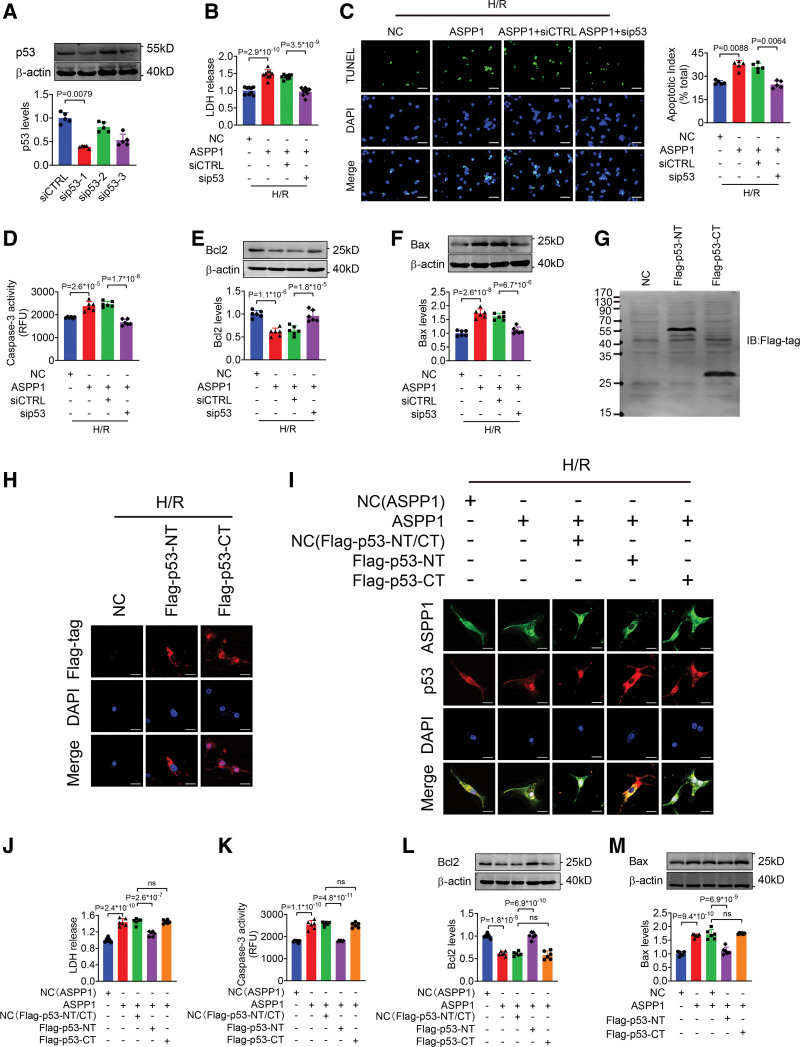
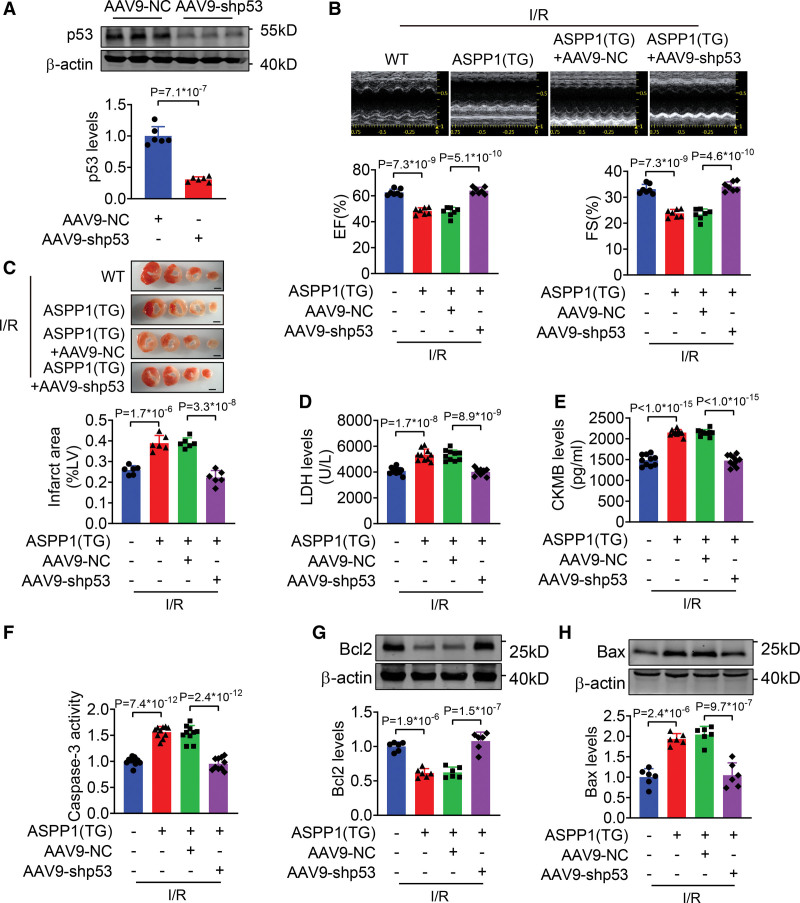
The immunofluorescent staining showed that under normal condition ASPP1 and p53 colocalized in the cytoplasm of neonatal mouse ventricular cardiomyocytes, while they were both upregulated and translocated to the nuclei upon hypoxia/reoxygenation treatment. The nuclear translocation of ASPP1 and p53 was interdependent, as knockdown of either ASPP1 or p53 attenuated nuclear translocation of the other one. Inhibition of importin-β1 resulted in the cytoplasmic sequestration of both p53 and ASPP1 in neonatal mouse ventricular cardiomyocytes with hypoxia/reoxygenation stimulation. Overexpression of ASPP1 potentiated, whereas knockdown of ASPP1 inhibited the expression of Bax (Bcl2-associated X), PUMA (p53 upregulated modulator of apoptosis), and Noxa, direct apoptosis-associated targets of p53. ASPP1 was also increased in the I/R myocardium. Cardiomyocyte-specific transgenic overexpression of ASPP1 aggravated I/R injury as indicated by increased infarct size and impaired cardiac function. Conversely, knockout of ASPP1 mitigated cardiac I/R injury. The same qualitative data were observed in neonatal mouse ventricular cardiomyocytes exposed to hypoxia/reoxygenation injury. Furthermore, inhibition of p53 significantly blunted the proapoptotic activity and detrimental effects of ASPP1 both in vitro and in vivo.
Binding of ASPP1 to p53 triggers their nuclear cotranslocation via importin-β1 that eventually exacerbates cardiac I/R injury. The findings imply that interfering the expression of ASPP1 or the interaction between ASPP1 and p53 to block their nuclear trafficking represents an important therapeutic strategy for cardiac I/R injury.
ASPP1(p53 蛋白凋亡刺激因子 1)作为 p53 的辅助因子,在调节细胞凋亡中起关键作用,可促进其在核内的转录活性。然而,细胞质 ASPP1 是否影响 p53 核易位及其在心脏疾病中的作用尚不清楚。本研究旨在探讨 ASPP1 调节 p53 核易位的机制及其随后对心肌缺血/再灌注(I/R)损伤的贡献。
免疫荧光染色显示,在正常情况下,ASPP1 和 p53 在新生鼠心室肌细胞的细胞质中共定位,而在低氧/复氧处理时,两者均上调并转位到核内。ASPP1 和 p53 的核易位是相互依赖的,因为敲低 ASPP1 或 p53 均可减弱另一种蛋白的核易位。抑制 importin-β1 导致新生鼠心室肌细胞在低氧/复氧刺激下 p53 和 ASPP1 滞留在细胞质中。ASPP1 的过表达增强了 p53 的表达,而 ASPP1 的敲低则抑制了 Bax(Bcl2 相关 X)、PUMA(p53 上调凋亡调节剂)和 Noxa(p53 的直接凋亡相关靶标)的表达。在 I/R 心肌中也增加了 ASPP1。ASPP1 的心肌细胞特异性转基因过表达加重了 I/R 损伤,表现为梗死面积增大和心功能受损。相反,ASPP1 的敲除减轻了心脏 I/R 损伤。在暴露于低氧/复氧损伤的新生鼠心室肌细胞中也观察到了相同的定性数据。此外,在体外和体内,抑制 p53 显著减弱了 ASPP1 的促凋亡活性和有害作用。
ASPP1 与 p53 的结合通过 importin-β1 触发它们的核共转位,最终加剧了心肌 I/R 损伤。这些发现表明,干扰 ASPP1 的表达或 ASPP1 与 p53 之间的相互作用以阻止其核易位,代表了治疗心肌 I/R 损伤的重要策略。