Alexanian Michael, Padmanabhan Arun, Nishino Tomohiro, Travers Joshua G, Ye Lin, Lee Clara Youngna, Sadagopan Nandhini, Huang Yu, Pelonero Angelo, Auclair Kirsten, Zhu Ada, Teran Barbara Gonzalez, Flanigan Will, Kim Charis Kee-Seon, Lumbao-Conradson Koya, Costa Mauro, Jain Rajan, Charo Israel, Haldar Saptarsi M, Pollard Katherine S, Vagnozzi Ronald J, McKinsey Timothy A, Przytycki Pawel F, Srivastava Deepak
Gladstone Institutes; San Francisco, CA, USA.
Roddenberry Center for Stem Cell Biology and Medicine at Gladstone Institutes; San Francisco, CA, USA.
bioRxiv. 2023 Jan 7:2023.01.06.522937. doi: 10.1101/2023.01.06.522937.
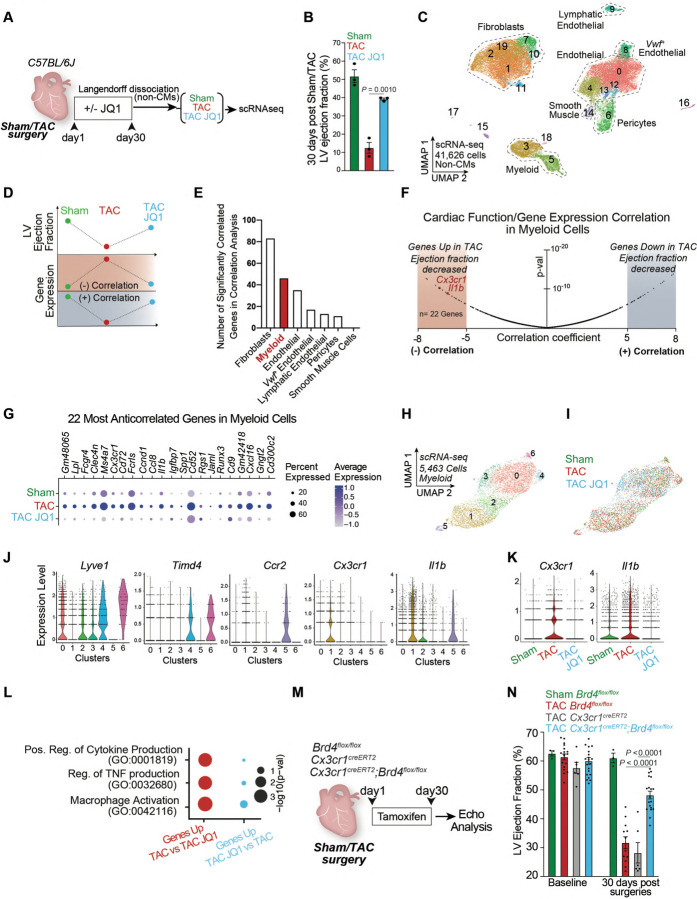
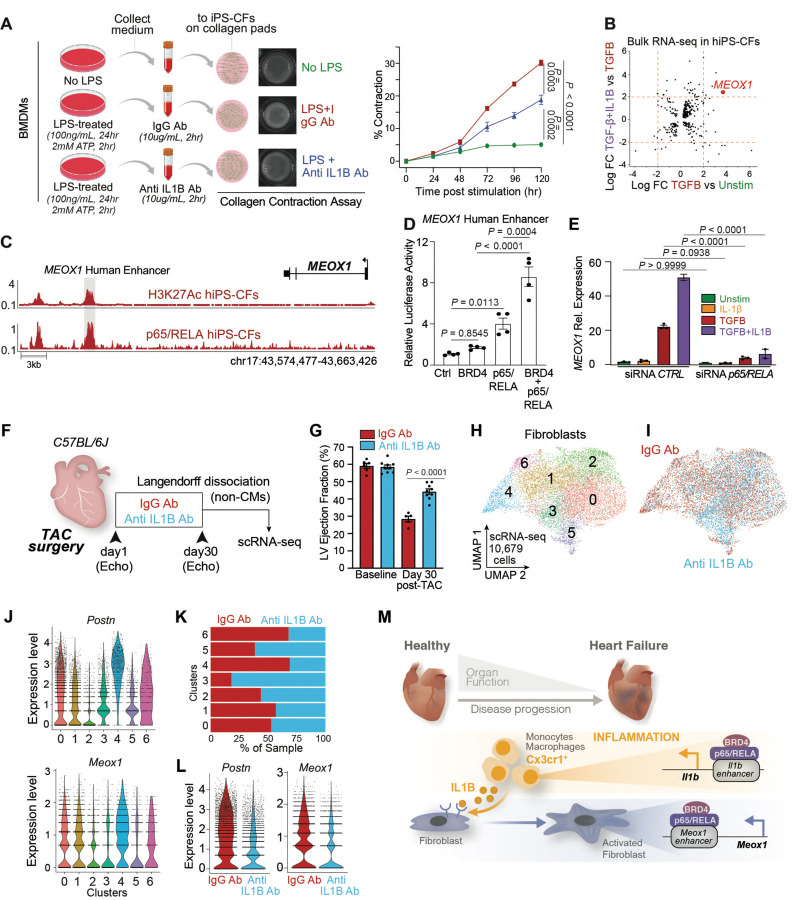
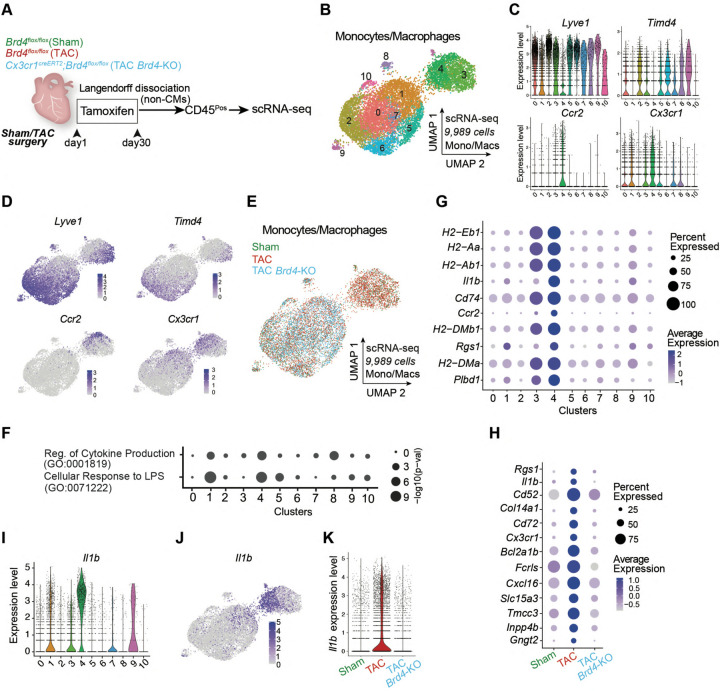
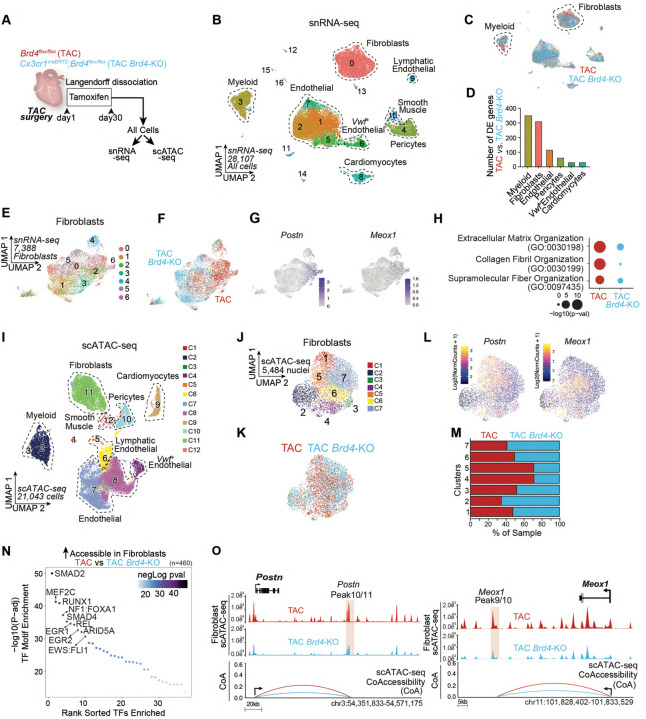
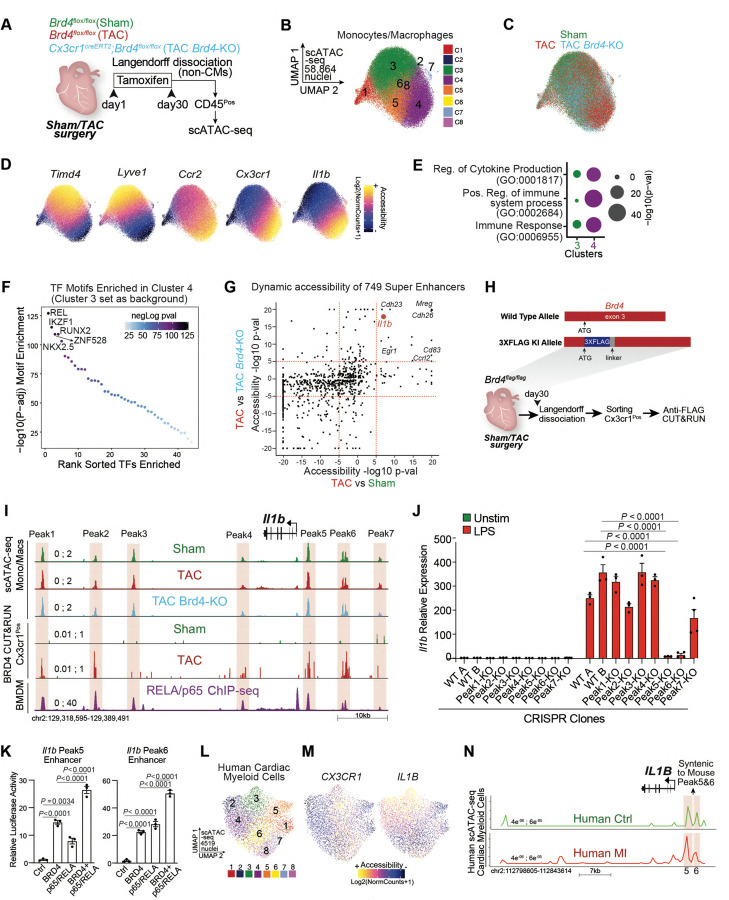
Chronic inflammation and tissue fibrosis are common stress responses that worsen organ function, yet the molecular mechanisms governing their crosstalk are poorly understood. In diseased organs, stress-induced changes in gene expression fuel maladaptive cell state transitions and pathological interaction between diverse cellular compartments. Although chronic fibroblast activation worsens dysfunction of lung, liver, kidney, and heart, and exacerbates many cancers, the stress-sensing mechanisms initiating the transcriptional activation of fibroblasts are not well understood. Here, we show that conditional deletion of the transcription co-activator in -positive myeloid cells ameliorates heart failure and is associated with a dramatic reduction in fibroblast activation. Analysis of single-cell chromatin accessibility and BRD4 occupancy in -positive cells identified a large enhancer proximal to (, and a series of CRISPR deletions revealed the precise stress-dependent regulatory element that controlled expression of in disease. Secreted IL1B functioned non-cell autonomously to activate a p65/RELA-dependent enhancer near the transcription factor , resulting in a profibrotic response in human cardiac fibroblasts. , antibody-mediated IL1B neutralization prevented stress-induced expression of , inhibited fibroblast activation, and improved cardiac function in heart failure. The elucidation of BRD4-dependent crosstalk between a specific immune cell subset and fibroblasts through IL1B provides new therapeutic strategies for heart disease and other disorders of chronic inflammation and maladaptive tissue remodeling.
慢性炎症和组织纤维化是常见的应激反应,会使器官功能恶化,但调控它们相互作用的分子机制却知之甚少。在患病器官中,应激诱导的基因表达变化推动了适应不良的细胞状态转变以及不同细胞区室之间的病理相互作用。尽管慢性成纤维细胞活化会加重肺、肝、肾和心脏的功能障碍,并加剧许多癌症,但启动成纤维细胞转录激活的应激感应机制尚不清楚。在这里,我们表明在γδ阳性髓样细胞中条件性缺失转录共激活因子可改善心力衰竭,并与成纤维细胞活化的显著减少相关。对γδ阳性细胞中单细胞染色质可及性和BRD4占据情况的分析确定了一个靠近[具体基因名称]的大型增强子([具体基因名称]),一系列CRISPR缺失揭示了在疾病中控制[具体基因名称]表达的精确应激依赖性调控元件。分泌的IL1B以非细胞自主方式发挥作用,激活转录因子[具体基因名称]附近的p65/RELA依赖性增强子,从而在人心脏成纤维细胞中引发促纤维化反应。[具体药物名称]抗体介导的IL1B中和可防止应激诱导的[具体基因名称]表达,抑制成纤维细胞活化,并改善心力衰竭中的心脏功能。通过IL1B阐明特定免疫细胞亚群与成纤维细胞之间依赖BRD4的相互作用,为心脏病以及慢性炎症和适应性不良组织重塑的其他疾病提供了新的治疗策略。