Department of Systems Biotechnology and Center for Antibiotic Resistome, Chung-Ang University, Anseong, 17546, Republic of Korea.
CJ Bioscience, Seoul, 04527, Republic of Korea.
Nat Commun. 2023 Mar 2;14(1):1191. doi: 10.1038/s41467-023-36633-7.
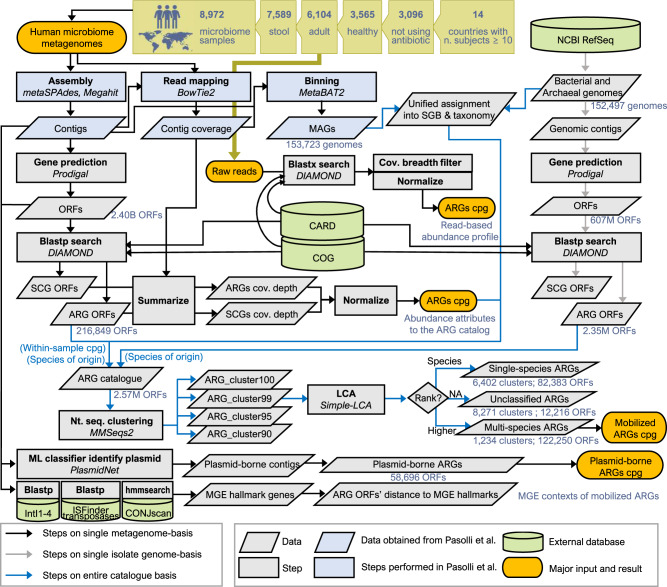
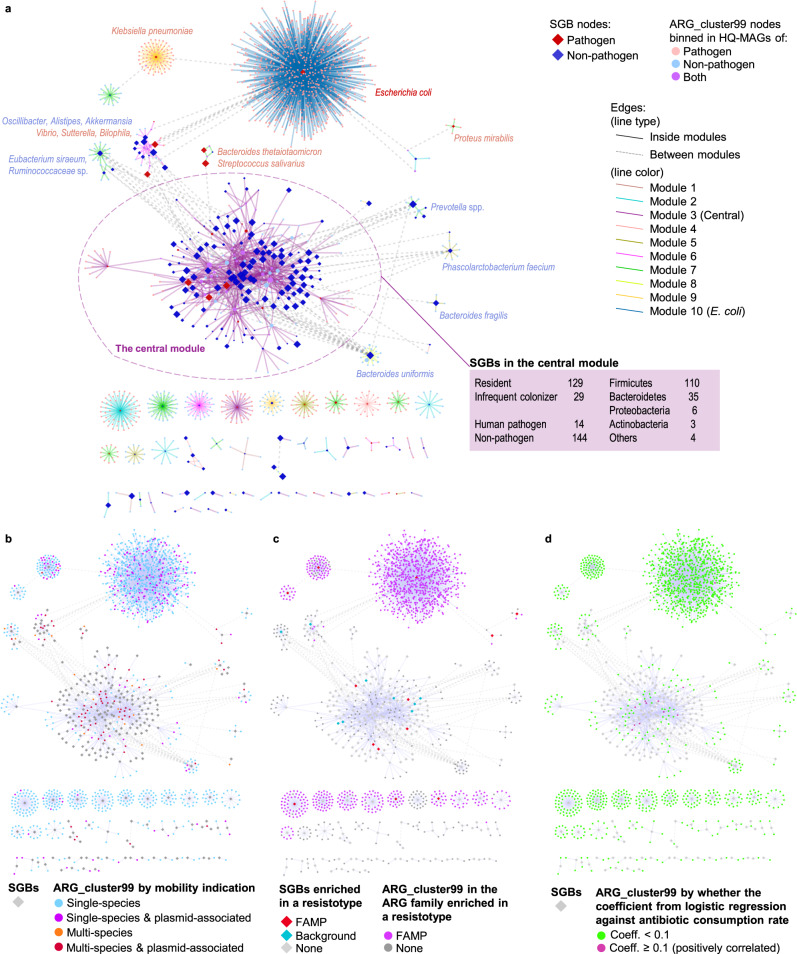
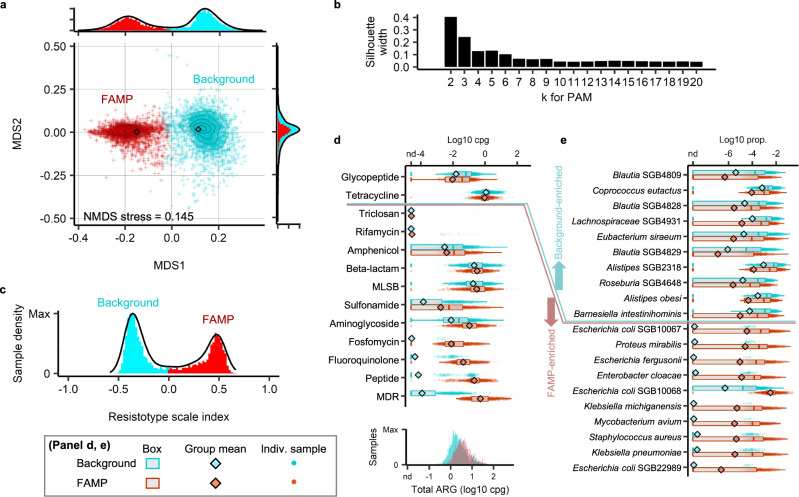
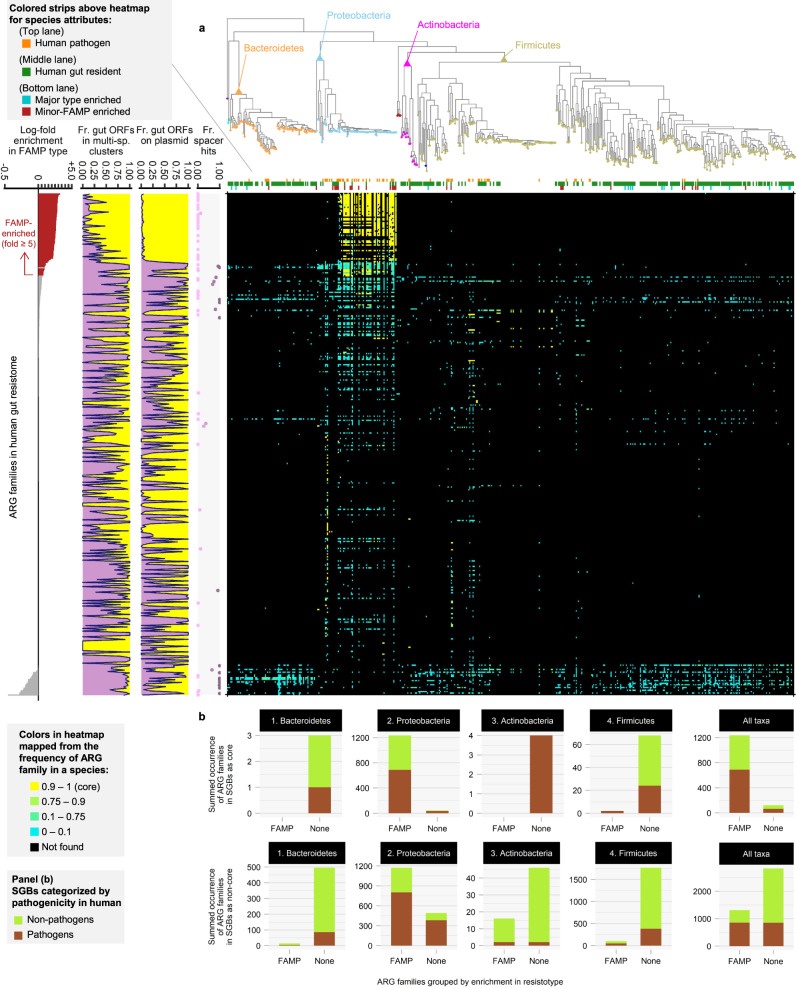
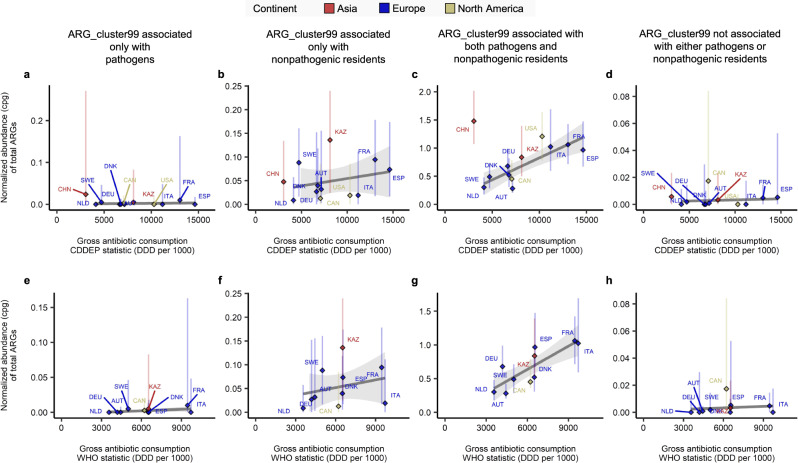
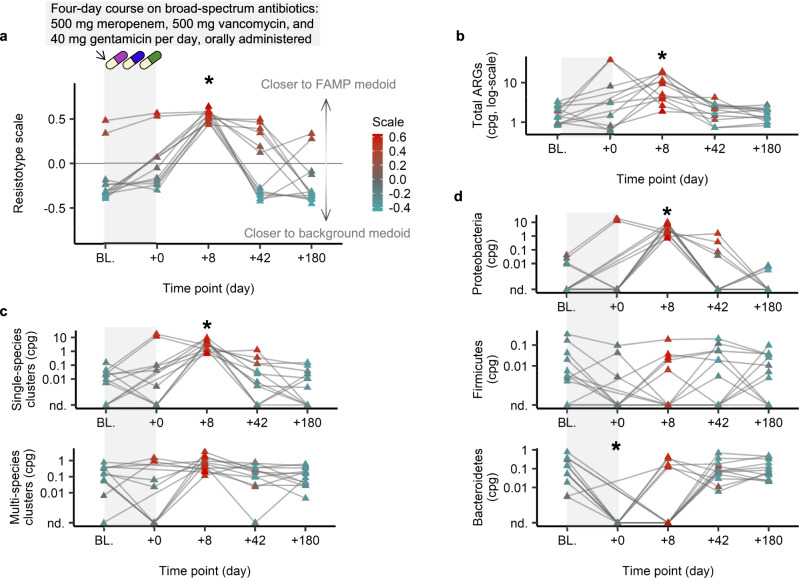
The widespread usage of antimicrobials has driven the evolution of resistance in pathogenic microbes, both increased prevalence of antimicrobial resistance genes (ARGs) and their spread across species by horizontal gene transfer (HGT). However, the impact on the wider community of commensal microbes associated with the human body, the microbiome, is less well understood. Small-scale studies have determined the transient impacts of antibiotic consumption but we conduct an extensive survey of ARGs in 8972 metagenomes to determine the population-level impacts. Focusing on 3096 gut microbiomes from healthy individuals not taking antibiotics we demonstrate highly significant correlations between both the total ARG abundance and diversity and per capita antibiotic usage rates across ten countries spanning three continents. Samples from China were notable outliers. We use a collection of 154,723 human-associated metagenome assembled genomes (MAGs) to link these ARGs to taxa and detect HGT. This reveals that the correlations in ARG abundance are driven by multi-species mobile ARGs shared between pathogens and commensals, within a highly connected central component of the network of MAGs and ARGs. We also observe that individual human gut ARG profiles cluster into two types or resistotypes. The less frequent resistotype has higher overall ARG abundance, is associated with certain classes of resistance, and is linked to species-specific genes in the Proteobacteria on the periphery of the ARG network.
抗生素的广泛使用推动了病原微生物耐药性的进化,包括抗菌药物耐药基因(ARGs)的普遍存在和通过水平基因转移(HGT)在物种间的传播。然而,人们对与人体共生的微生物群落(微生物组)的广泛影响了解较少。小规模研究已经确定了抗生素消费的短暂影响,但我们对 8972 个宏基因组进行了广泛的 ARG 调查,以确定其对人群的影响。我们专注于来自未服用抗生素的 3096 个健康个体的肠道微生物组,证明了在跨越三大洲的十个国家中,总 ARG 丰度和多样性以及人均抗生素使用率之间存在高度显著的相关性。来自中国的样本是明显的异常值。我们使用了 154723 个人类相关宏基因组组装基因组(MAGs)的集合,将这些 ARG 与分类群联系起来,并检测 HGT。这表明,ARG 丰度的相关性是由病原体和共生菌之间共享的多物种移动 ARG 驱动的,这些移动 ARG 位于 MAG 和 ARG 网络的一个高度连接的中心组件内。我们还观察到,个体人类肠道 ARG 谱聚类为两种类型或耐药型。较少见的耐药型具有更高的总体 ARG 丰度,与某些类别的耐药性相关,并且与 ARG 网络边缘的变形菌门的特定种属基因有关。