Yang Xin, Bhowmik Arghya, Vegge Tejs, Hansen Heine Anton
Department of Energy Conversion and Storage, Technical University of Denmark Anker Engelunds Vej, 2800 Kgs Lyngby Denmark
Chem Sci. 2023 Mar 13;14(14):3913-3922. doi: 10.1039/d2sc06696c. eCollection 2023 Apr 5.
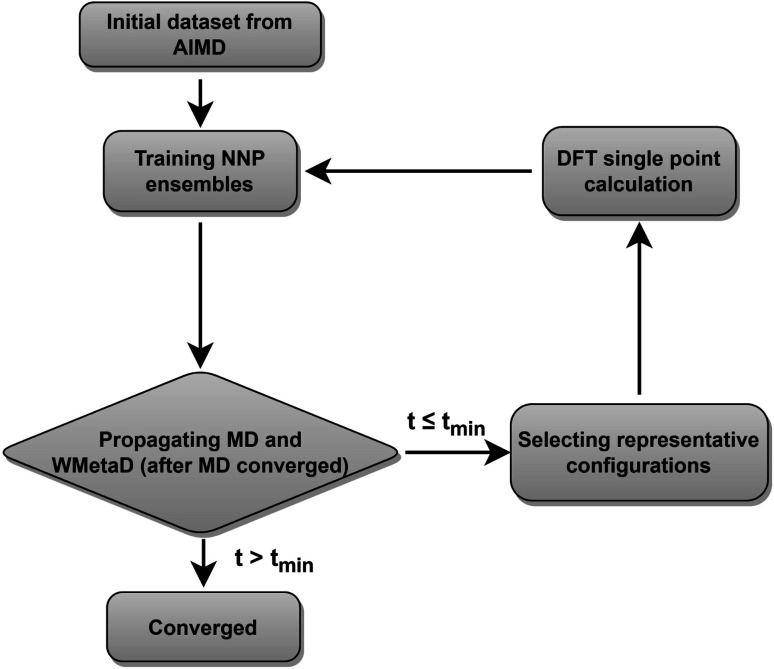
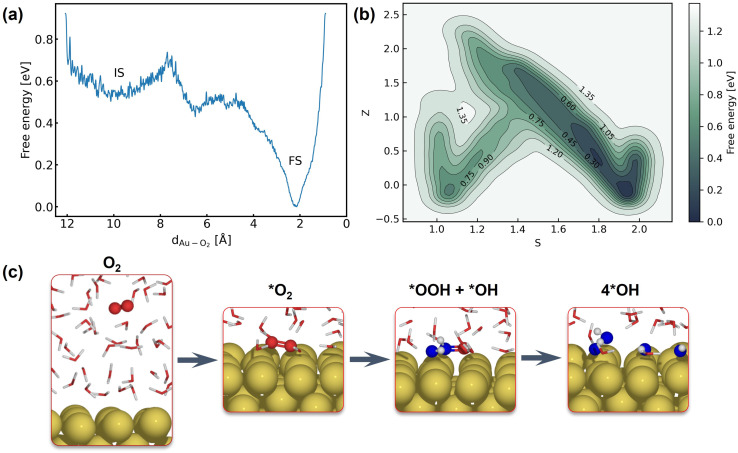
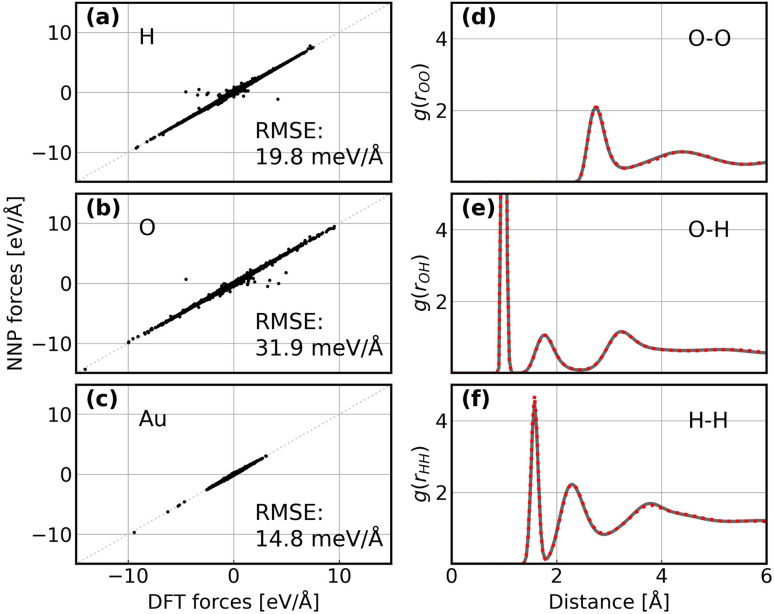
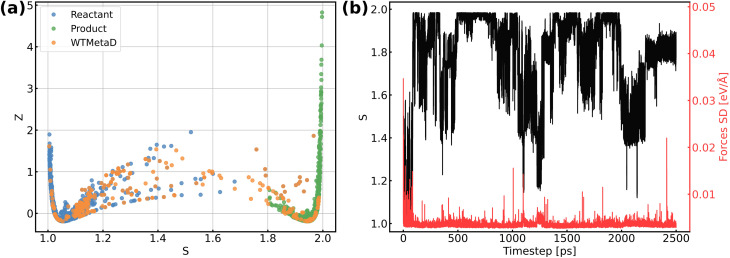
The application of molecular dynamics (AIMD) for the explicit modeling of reactions at solid-liquid interfaces in electrochemical energy conversion systems like batteries and fuel cells can provide new understandings towards reaction mechanisms. However, its prohibitive computational cost severely restricts the time- and length-scales of AIMD. Equivariant graph neural network (GNN) based accurate surrogate potentials can accelerate the speed of performing molecular dynamics after learning on representative structures in a data efficient manner. In this study, we combined uncertainty-aware GNN potentials and enhanced sampling to investigate the reactive process of the oxygen reduction reaction (ORR) at an Au(100)-water interface. By using a well-established active learning framework based on CUR matrix decomposition, we can evenly sample equilibrium structures from MD simulations and non-equilibrium reaction intermediates that are rarely visited during the reaction. The trained GNNs have shown exceptional performance in terms of force prediction accuracy, the ability to reproduce structural properties, and low uncertainties when performing MD and metadynamics simulations. Furthermore, the collective variables employed in this work enabled the automatic search of reaction pathways and provide a detailed understanding towards the ORR reaction mechanism on Au(100). Our simulations identified the associative reaction mechanism without the presence of *O and a low reaction barrier of 0.3 eV, which is in agreement with experimental findings. The methodology employed in this study can pave the way for modeling complex chemical reactions at electrochemical interfaces with an explicit solvent under ambient conditions.
在电池和燃料电池等电化学能量转换系统中,将分子动力学(AIMD)应用于固液界面反应的显式建模,可为反应机理提供新的认识。然而,其高昂的计算成本严重限制了AIMD的时间和长度尺度。基于等变图神经网络(GNN)的精确替代势能够以数据高效的方式在代表性结构上进行学习后,加快分子动力学的执行速度。在本研究中,我们结合了不确定性感知GNN势和增强采样,以研究Au(100)-水界面处氧还原反应(ORR)的反应过程。通过使用基于CUR矩阵分解的成熟主动学习框架,我们可以从MD模拟中均匀采样平衡结构以及反应过程中很少出现的非平衡反应中间体。经过训练的GNN在力预测精度、再现结构性质的能力以及进行MD和元动力学模拟时的低不确定性方面表现出色。此外,本工作中使用的集体变量能够自动搜索反应路径,并对Au(100)上的ORR反应机理提供详细理解。我们的模拟确定了不存在*O的缔合反应机理以及0.3 eV的低反应势垒,这与实验结果一致。本研究中采用的方法可为在环境条件下用显式溶剂对电化学界面处的复杂化学反应进行建模铺平道路。