Lambisia Arnold W, Makori Timothy O, Mutunga Martin, Cheruiyot Robinson, Murunga Nickson, Quick Joshua, Githinji George, Nokes D James, Houldcroft Charlotte J, Agoti Charles N
Kenya Medical Research Institute-Wellcome Trust Research Programme, PO Box 230-80108, Kilifi, Kenya.
Institute of Microbiology and Infection, School of Biosciences, University of Birmingham, Birmingham B15 2TT, UK.
Virus Evol. 2023 Mar 24;9(1):vead023. doi: 10.1093/ve/vead023. eCollection 2023.
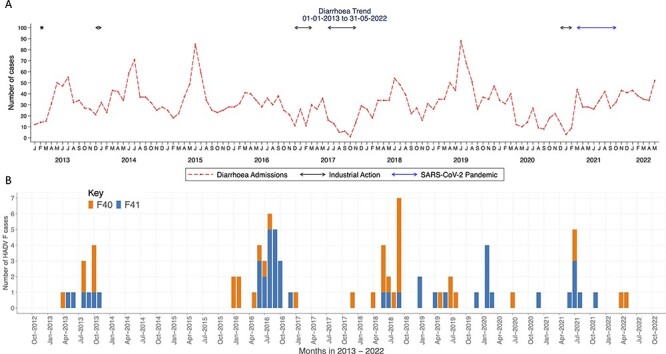
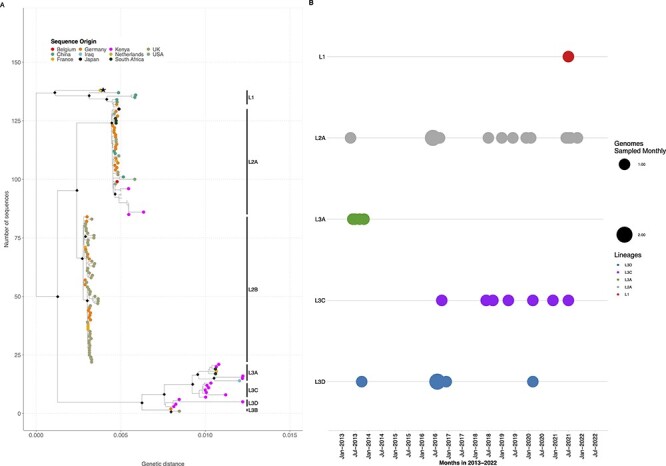
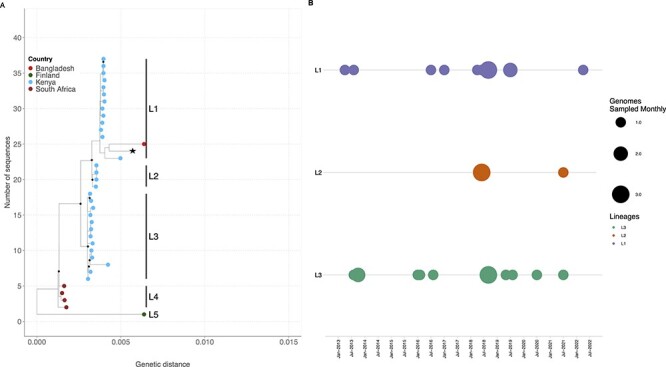
Human enteric adenovirus species F (HAdV-F) is a leading cause of childhood diarrhoeal deaths. The genomic analysis would be key to understanding transmission dynamics, potential drivers of disease severity, and vaccine development. However, currently, there are limited HAdV-F genomic data globally. Here, we sequenced and analysed HAdV-F from stool samples collected in coastal Kenya between 2013 and 2022. The samples were collected at Kilifi County Hospital in coastal Kenya from children <13 years of age who reported a history of three or more loose stools in the previous 24 hours. The genomes were analysed together with the data from the rest of the world by phylogenetic analysis and mutational profiling. Types and lineages were assigned based on phylogenetic clustering consistent with the previously described criteria and nomenclature. Participant clinical and demographic data were linked to genotypic data. Of ninety-one cases identified using real-time Polymerase Chain Reaction, eighty-eight near-complete genomes were assembled, and these were classified into HAdV-F40 ( = 41) and HAdV-F41 ( = 47). These types co-circulated throughout the study period. Three and four distinct lineages were observed for HAdV-F40 (Lineages 1-3) and HAdV-F41 (Lineages 1, 2A, 3A, 3C, and 3D). Types F40 and F41 coinfections were observed in five samples and F41 and B7 in one sample. Two children with F40 and 41 coinfections were also infected with rotavirus and had moderate and severe diseases as defined using the Vesikari Scoring System, respectively. Intratypic recombination was found in four HAdV-F40 sequences occurring between Lineages 1 and 3. None of the HAdV-F41 cases had jaundice. This study provides evidence of extensive genetic diversity, coinfections, and recombination within HAdV-F40 in a rural coastal Kenya that will inform public health policy, vaccine development that includes the locally circulating lineages, and molecular diagnostic assay development. We recommend future comprehensive studies elucidating on HAdV-F genetic diversity and immunity for rational vaccine development.
人类肠道腺病毒F种(HAdV-F)是导致儿童腹泻死亡的主要原因。基因组分析对于理解传播动态、疾病严重程度的潜在驱动因素以及疫苗开发至关重要。然而,目前全球范围内HAdV-F的基因组数据有限。在此,我们对2013年至2022年间在肯尼亚沿海地区收集的粪便样本中的HAdV-F进行了测序和分析。样本是在肯尼亚沿海基利菲县医院从13岁以下儿童中采集的,这些儿童报告在过去24小时内有三次或更多次稀便病史。通过系统发育分析和突变谱分析,将这些基因组与来自世界其他地区的数据一起进行分析。根据与先前描述的标准和命名法一致的系统发育聚类来确定类型和谱系。将参与者的临床和人口统计学数据与基因型数据相关联。在使用实时聚合酶链反应鉴定出的91例病例中,组装了88个近乎完整的基因组,这些基因组被分类为HAdV-F40(=41)和HAdV-F41(=47)。在整个研究期间,这些类型共同流行。观察到HAdV-F40(谱系1-3)和HAdV-F41(谱系1、2A、3A、3C和3D)分别有三个和四个不同的谱系。在五个样本中观察到F40和F41型共感染,在一个样本中观察到F41和B7型共感染。两名同时感染F40和41型的儿童也感染了轮状病毒,分别患有使用韦西卡里评分系统定义的中度和重度疾病。在谱系1和3之间的四个HAdV-F40序列中发现了型内重组。所有HAdV-F41病例均无黄疸。这项研究提供了证据,证明肯尼亚沿海农村地区的HAdV-F40存在广泛的遗传多样性、共感染和重组,这将为公共卫生政策、包括当地流行谱系的疫苗开发以及分子诊断检测方法的开发提供信息。我们建议未来进行全面研究,阐明HAdV-F的遗传多样性和免疫情况,以推动合理的疫苗开发。