Institute of Crop Sciences, Chinese Academy of Agricultural Sciences/National Engineering Laboratory for Crop Molecular Breeding, National Center of Space Mutagenesis for Crop Improvement, Beijing, China.
Biophysics Group, Institute of Modern Physics, Chinese Academy of Sciences, Lanzhou, China.
Plant Biotechnol J. 2023 Oct;21(10):2047-2056. doi: 10.1111/pbi.14111. Epub 2023 Jul 3.
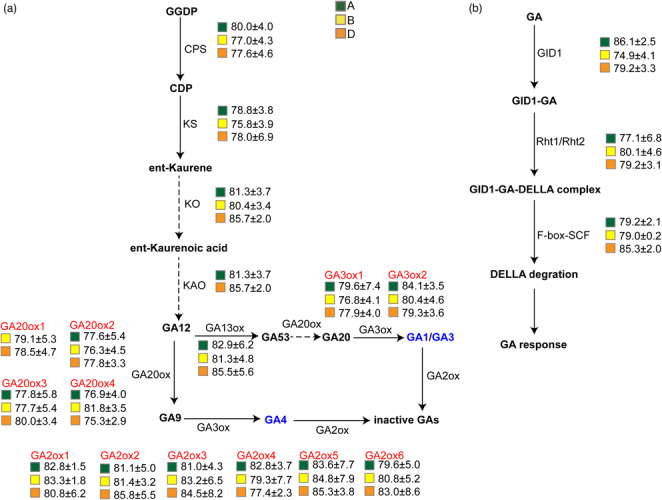
Hexaploid wheat (Triticum aestivum), a major staple crop, has a remarkably large genome of ~14.4 Gb (containing 106 913 high-confidence [HC] and 159 840 low-confidence [LC] genes in the Chinese Spring v2.1 reference genome), which poses a major challenge for functional genomics studies. To overcome this hurdle, we performed whole-exome sequencing to generate a nearly saturated wheat mutant database containing 18 025 209 mutations induced by ethyl methanesulfonate (EMS), carbon (C)-ion beams, or γ-ray mutagenesis. This database contains an average of 47.1 mutations per kb in each gene-coding sequence: the potential functional mutations were predicted to cover 96.7% of HC genes and 70.5% of LC genes. Comparative analysis of mutations induced by EMS, γ-rays, or C-ion beam irradiation revealed that γ-ray and C-ion beam mutagenesis induced a more diverse array of variations than EMS, including large-fragment deletions, small insertions/deletions, and various non-synonymous single nucleotide polymorphisms. As a test case, we combined mutation analysis with phenotypic screening and rapidly mapped the candidate gene responsible for the phenotype of a yellow-green leaf mutant to a 2.8-Mb chromosomal region. Furthermore, a proof-of-concept reverse genetics study revealed that mutations in gibberellic acid biosynthesis and signalling genes could be associated with negative impacts on plant height. Finally, we built a publically available database of these mutations with the corresponding germplasm (seed stock) repository to facilitate advanced functional genomics studies in wheat for the broad plant research community.
六倍体小麦(Triticum aestivum)是一种主要的主食作物,其基因组非常庞大,约为 144 亿碱基对(中国春 v2.1 参考基因组中包含 106913 个高可信度 [HC]和 159840 个低可信度 [LC]基因),这对功能基因组学研究构成了重大挑战。为了克服这一障碍,我们进行了全外显子组测序,生成了一个几乎饱和的小麦突变体数据库,其中包含了 18025209 个由乙基甲磺酸(EMS)、碳(C)离子束或γ射线诱变诱导的突变。该数据库中每个基因编码序列的平均突变数为 47.1 个/ kb:预测潜在功能突变将覆盖 96.7%的 HC 基因和 70.5%的 LC 基因。对 EMS、γ射线和 C 离子束诱变诱导的突变进行比较分析表明,γ射线和 C 离子束诱变比 EMS 诱导产生了更多种类的变异,包括大片段缺失、小插入/缺失和各种非同义单核苷酸多态性。作为一个测试案例,我们将突变分析与表型筛选相结合,快速将一个黄绿叶突变体的候选基因定位到一个 2.8 Mb 的染色体区域。此外,一项概念验证的反向遗传学研究表明,赤霉素生物合成和信号转导基因的突变可能与植株高度的负面影响有关。最后,我们建立了一个公共的突变数据库,其中包含相应的种质(种子库存)库,以方便广大植物研究界在小麦中进行先进的功能基因组学研究。