Neugebauer Hagen, Pinski Peter, Grimme Stefan, Neese Frank, Bursch Markus
Mulliken Center for Theoretical Chemistry, Clausius Institute for Physical and Theoretical Chemistry, University of Bonn, Beringstraße 4, D-53115 Bonn, Germany.
HQS Quantum Simulations GmbH, Rintheimer Straße 23, D-76131 Karlsruhe, Germany.
J Chem Theory Comput. 2023 Nov 14;19(21):7695-7703. doi: 10.1021/acs.jctc.3c00896. Epub 2023 Oct 20.
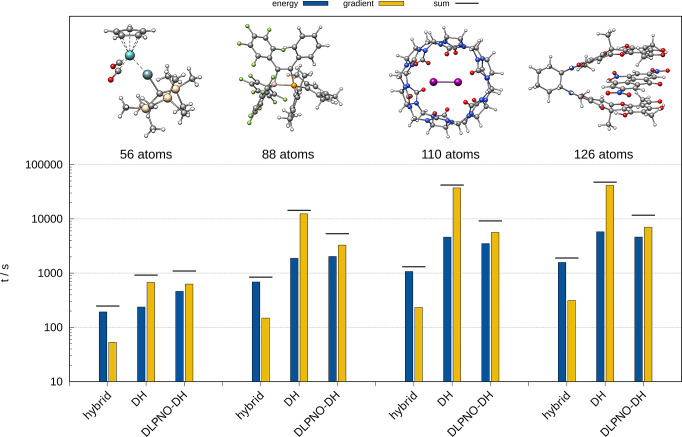
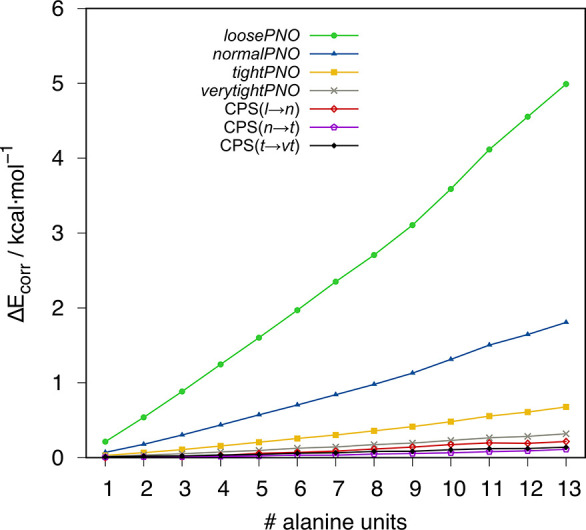
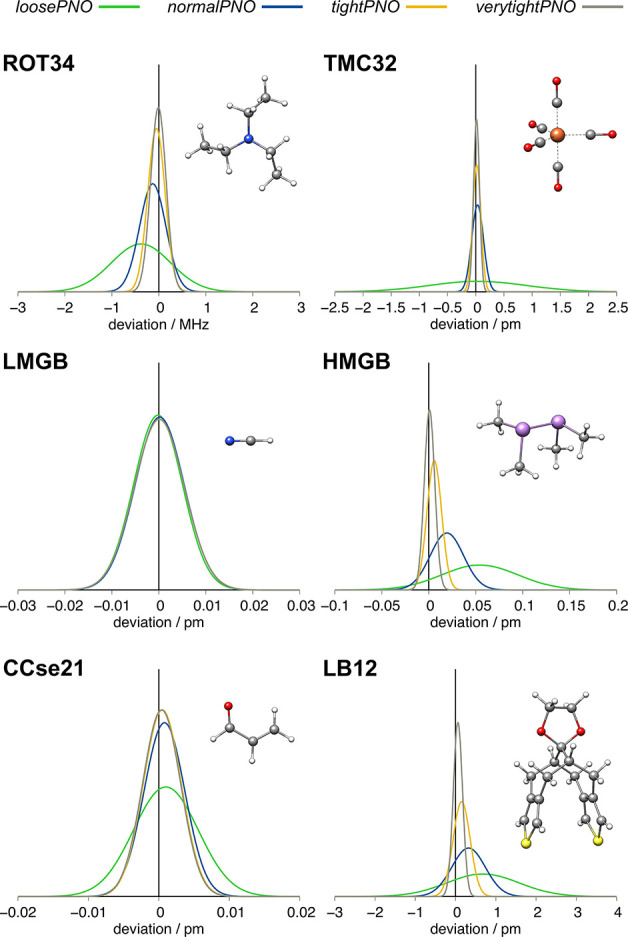
The unfavorable scaling () of the conventional second-order Møller-Plesset theory (MP2) typically prevents the application of double-hybrid (DH) density functionals to large systems with more than 100 atoms. A prominent approach to reduce the computational demand of electron correlation methods is the domain-based local pair natural orbital (DLPNO) approximation that is successfully used in the framework of DLPNO-CCSD(T). Its extension to MP2 [Pinski P.; Riplinger, C.; Valeev, E. F.; Neese, F. , , 034108.] paved the way for DLPNO-based DH (DLPNO-DH) methods. In this work, we assess the accuracy of the DLPNO-DH approximation compared to conventional DHs on a large number of 7925 data points for thermochemistry and 239 data points for structural features, including main-group and transition-metal systems. It is shown that DLPNO-DH-DFT can be applied successfully to perform energy calculations and geometry optimizations for large molecules at a drastically reduced computational cost. Furthermore, PNO space extrapolation is shown to be applicable, similar to its DLPNO-CCSD(T) counterpart, to reduce the remaining error.
传统二阶莫勒-普莱塞特理论(MP2)的不利标度通常阻碍了双杂化(DH)密度泛函在含有100多个原子的大体系中的应用。降低电子相关方法计算需求的一种突出方法是基于域的定域对自然轨道(DLPNO)近似,它在DLPNO-CCSD(T)框架中得到了成功应用。将其扩展到MP2 [Pinski P.; Riplinger, C.; Valeev, E. F.; Neese, F.,, 034108.] 为基于DLPNO的DH(DLPNO-DH)方法铺平了道路。在这项工作中,我们在大量包含7925个热化学数据点和239个结构特征数据点(包括主族和过渡金属体系)上,评估了DLPNO-DH近似与传统DH相比的准确性。结果表明,DLPNO-DH-DFT能够以大幅降低的计算成本成功应用于大分子的能量计算和几何结构优化。此外,与DLPNO-CCSD(T)类似,PNO空间外推法也可用于减少剩余误差。