Department of Medical Microbiology, UMC Utrecht, Utrecht, The Netherlands.
Utrecht University, Institute for Risk Assessment Sciences, Utrecht, The Netherlands.
PeerJ. 2024 Jan 4;12:e16695. doi: 10.7717/peerj.16695. eCollection 2024.
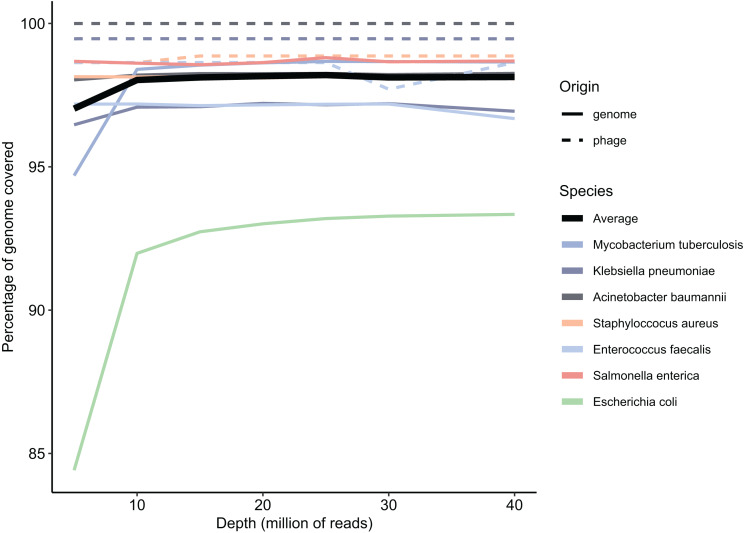
Antimicrobial resistance genes (ARG) are commonly found on acquired mobile genetic elements (MGEs) such as plasmids or transposons. Understanding the spread of resistance genes associated with mobile elements (mARGs) across different hosts and environments requires linking ARGs to the existing mobile reservoir within bacterial communities. However, reconstructing mARGs in metagenomic data from diverse ecosystems poses computational challenges, including genome fragment reconstruction (assembly), high-throughput annotation of MGEs, and identification of their association with ARGs. Recently, several bioinformatics tools have been developed to identify assembled fragments of plasmids, phages, and insertion sequence (IS) elements in metagenomic data. These methods can help in understanding the dissemination of mARGs. To streamline the process of identifying mARGs in multiple samples, we combined these tools in an automated high-throughput open-source pipeline, MetaMobilePicker, that identifies ARGs associated with plasmids, IS elements and phages, starting from short metagenomic sequencing reads. This pipeline was used to identify these three elements on a simplified simulated metagenome dataset, comprising whole genome sequences from seven clinically relevant bacterial species containing 55 ARGs, nine plasmids and five phages. The results demonstrated moderate precision for the identification of plasmids (0.57) and phages (0.71), and moderate sensitivity of identification of IS elements (0.58) and ARGs (0.70). In this study, we aim to assess the main causes of this moderate performance of the MGE prediction tools in a comprehensive manner. We conducted a systematic benchmark, considering metagenomic read coverage, contig length cutoffs and investigating the performance of the classification algorithms. Our analysis revealed that the metagenomic assembly process is the primary bottleneck when linking ARGs to identified MGEs in short-read metagenomics sequencing experiments rather than ARGs and MGEs identification by the different tools.
抗微生物耐药基因(ARG)通常存在于获得性移动遗传元件(MGE)上,例如质粒或转座子。了解与移动元件(mARGs)相关的耐药基因在不同宿主和环境中的传播,需要将 ARGs 与细菌群落中现有的移动库联系起来。然而,在来自不同生态系统的宏基因组数据中重建 mARGs 存在计算挑战,包括基因组片段重建(组装)、MGE 的高通量注释以及识别它们与 ARGs 的关联。最近,已经开发了几种生物信息学工具来识别宏基因组数据中质粒、噬菌体和插入序列(IS)元件的组装片段。这些方法有助于理解 mARGs 的传播。为了简化在多个样本中识别 mARGs 的过程,我们将这些工具组合在一个自动化的高通量开源管道 MetaMobilePicker 中,该管道从短的宏基因组测序reads 开始,识别与质粒、IS 元件和噬菌体相关的 ARGs。该管道用于在简化的模拟宏基因组数据集上识别这三个元素,该数据集包含七个具有 55 个 ARGs、九个质粒和五个噬菌体的临床相关细菌物种的全基因组序列。结果表明,质粒(0.57)和噬菌体(0.71)的识别具有中等精度,IS 元件(0.58)和 ARGs(0.70)的识别具有中等敏感性。在这项研究中,我们旨在全面评估 MGE 预测工具这种中等性能的主要原因。我们进行了系统的基准测试,考虑了宏基因组读覆盖率、contig 长度截止值,并研究了分类算法的性能。我们的分析表明,在短读宏基因组测序实验中,将 ARGs 与鉴定的 MGEs 联系起来的主要瓶颈是宏基因组组装过程,而不是不同工具对 ARGs 和 MGEs 的鉴定。