Zhu Bokai, Gao Sheng, Chen Shuxiao, Yeung Jason, Bai Yunhao, Huang Amy Y, Yeo Yao Yu, Liao Guanrui, Mao Shulin, Jiang Zhenghui G, Rodig Scott J, Shalek Alex K, Nolan Garry P, Jiang Sizun, Ma Zongming
Ragon Institute of MGH, MIT, and Harvard, Cambridge, MA, USA.
Broad Institute of MIT and Harvard, Cambridge, MA, USA.
bioRxiv. 2024 May 14:2024.05.12.593710. doi: 10.1101/2024.05.12.593710.
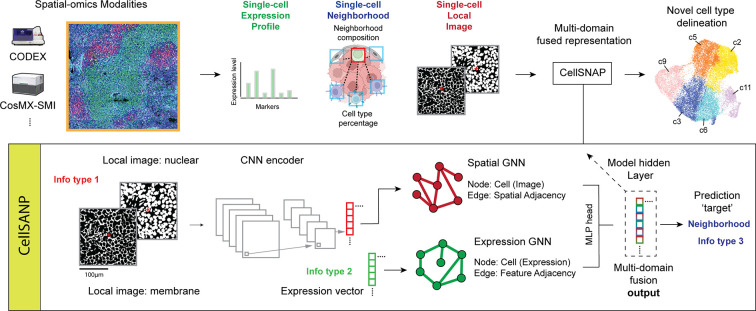
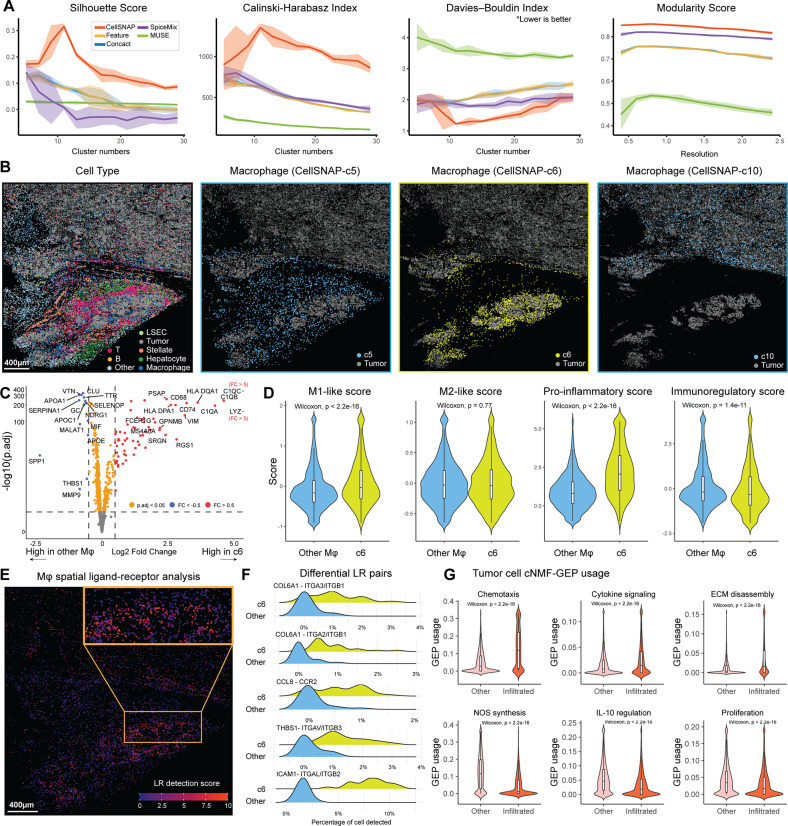
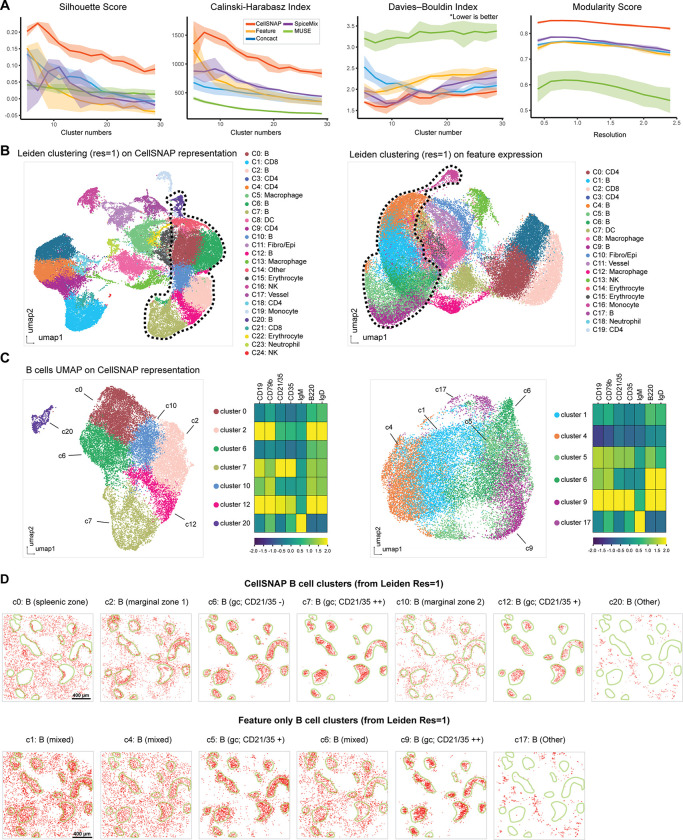
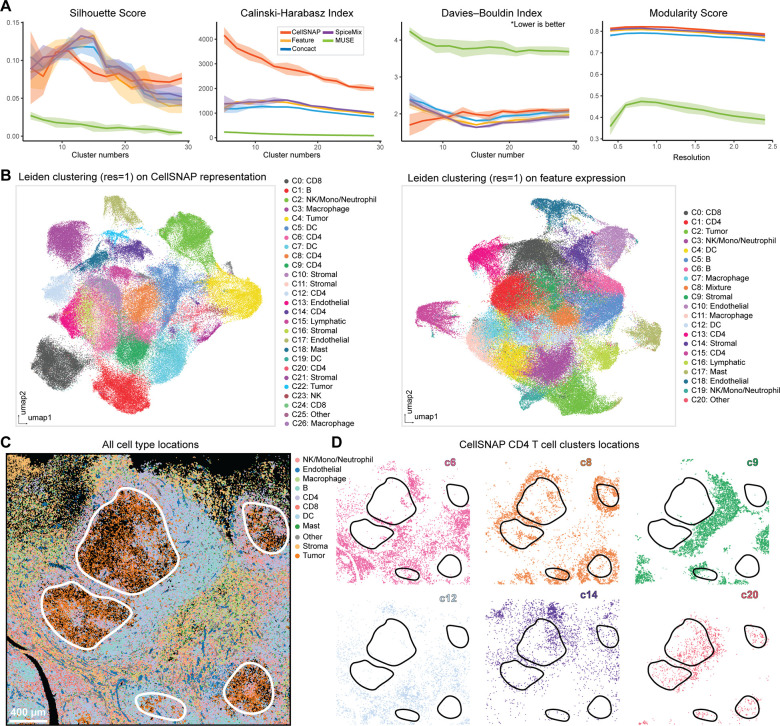
Cell population delineation and identification is an essential step in single-cell and spatial-omics studies. Spatial-omics technologies can simultaneously measure information from three complementary domains related to this task: expression levels of a panel of molecular biomarkers at single-cell resolution, relative positions of cells, and images of tissue sections, but existing computational methods for performing this task on single-cell spatial-omics datasets often relinquish information from one or more domains. The additional reliance on the availability of "atlas" training or reference datasets limits cell type discovery to well-defined but limited cell population labels, thus posing major challenges for using these methods in practice. Successful integration of all three domains presents an opportunity for uncovering cell populations that are functionally stratified by their spatial contexts at cellular and tissue levels: the key motivation for employing spatial-omics technologies in the first place. In this work, we introduce Cell Spatio- and Neighborhood-informed Annotation and Patterning (CellSNAP), a self-supervised computational method that learns a representation vector for each cell in tissue samples measured by spatial-omics technologies at the single-cell or finer resolution. The learned representation vector fuses information about the corresponding cell across all three aforementioned domains. By applying CellSNAP to datasets spanning both spatial proteomic and spatial transcriptomic modalities, and across different tissue types and disease settings, we show that CellSNAP markedly enhances discovery of biologically relevant cell populations at fine granularity, beyond current approaches, by fully integrating cells' molecular profiles with cellular neighborhood and tissue image information.
细胞群体的划分和识别是单细胞和空间组学研究中的关键步骤。空间组学技术能够同时测量与该任务相关的三个互补领域的信息:单细胞分辨率下一组分子生物标志物的表达水平、细胞的相对位置以及组织切片图像。然而,现有的用于在单细胞空间组学数据集上执行此任务的计算方法往往会舍弃一个或多个领域的信息。对“图谱”训练或参考数据集可用性的额外依赖,将细胞类型的发现限制在定义明确但有限的细胞群体标签上,这给在实际中使用这些方法带来了重大挑战。成功整合所有这三个领域为揭示在细胞和组织水平上按空间背景进行功能分层的细胞群体提供了机会,这也是最初采用空间组学技术的关键动机。在这项工作中,我们引入了细胞空间和邻域信息注释与模式化方法(CellSNAP),这是一种自监督计算方法,可在单细胞或更精细分辨率下为通过空间组学技术测量的组织样本中的每个细胞学习一个表示向量。所学习的表示向量融合了上述所有三个领域中关于相应细胞的信息。通过将CellSNAP应用于跨越空间蛋白质组学和空间转录组学模式、不同组织类型和疾病背景的数据集,我们表明,通过将细胞的分子谱与细胞邻域和组织图像信息充分整合,CellSNAP显著增强了在精细粒度上发现生物学相关细胞群体的能力,超越了现有方法。