Misra Sima, Crosby Madeline A, Mungall Christopher J, Matthews Beverley B, Campbell Kathryn S, Hradecky Pavel, Huang Yanmei, Kaminker Joshua S, Millburn Gillian H, Prochnik Simon E, Smith Christopher D, Tupy Jonathan L, Whitfied Eleanor J, Bayraktaroglu Leyla, Berman Benjamin P, Bettencourt Brian R, Celniker Susan E, de Grey Aubrey D N J, Drysdale Rachel A, Harris Nomi L, Richter John, Russo Susan, Schroeder Andrew J, Shu Sheng Qiang, Stapleton Mark, Yamada Chihiro, Ashburner Michael, Gelbart William M, Rubin Gerald M, Lewis Suzanna E
Department of Molecular and Cell Biology, University of California, Life Sciences Addition, Berkeley, CA 94720-3200, USA.
Genome Biol. 2002;3(12):RESEARCH0083. doi: 10.1186/gb-2002-3-12-research0083. Epub 2002 Dec 31.
The recent completion of the Drosophila melanogaster genomic sequence to high quality and the availability of a greatly expanded set of Drosophila cDNA sequences, aligning to 78% of the predicted euchromatic genes, afforded FlyBase the opportunity to significantly improve genomic annotations. We made the annotation process more rigorous by inspecting each gene visually, utilizing a comprehensive set of curation rules, requiring traceable evidence for each gene model, and comparing each predicted peptide to SWISS-PROT and TrEMBL sequences.
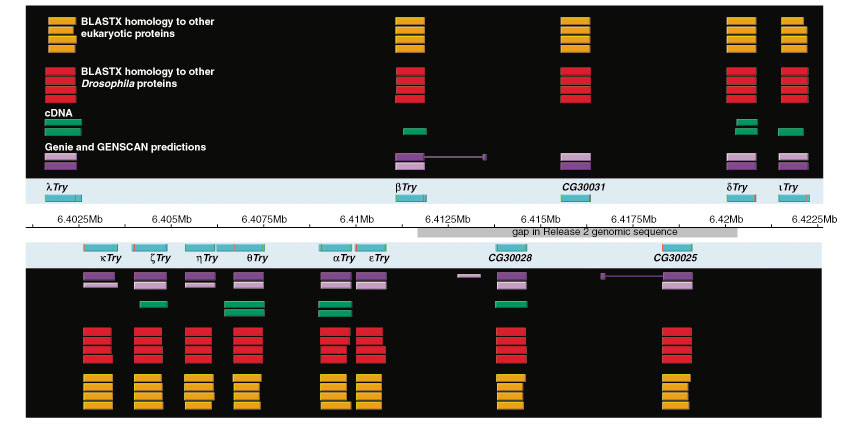
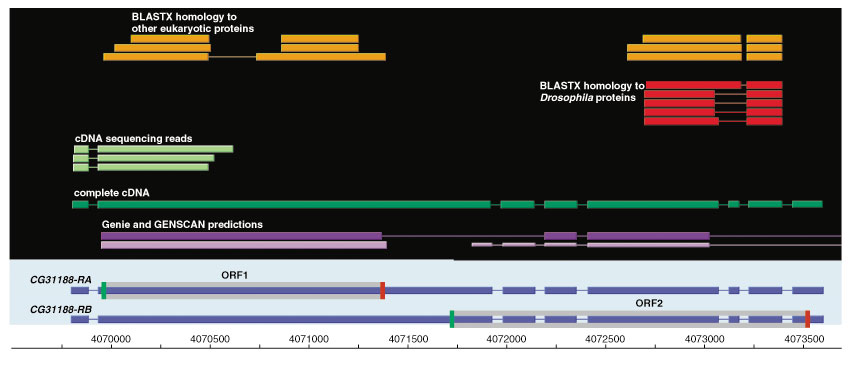
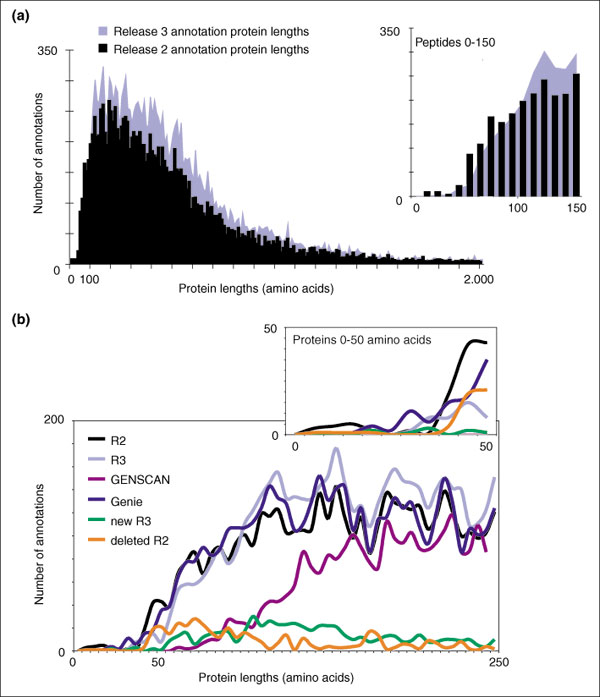
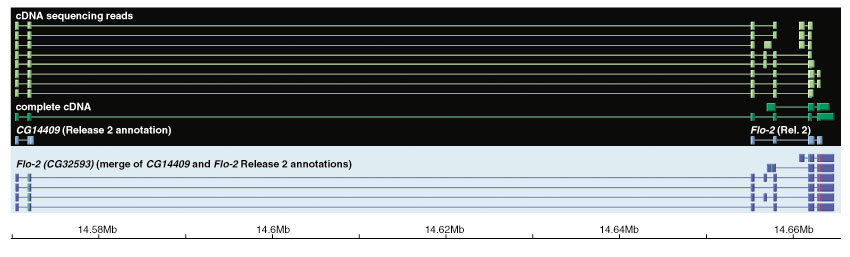
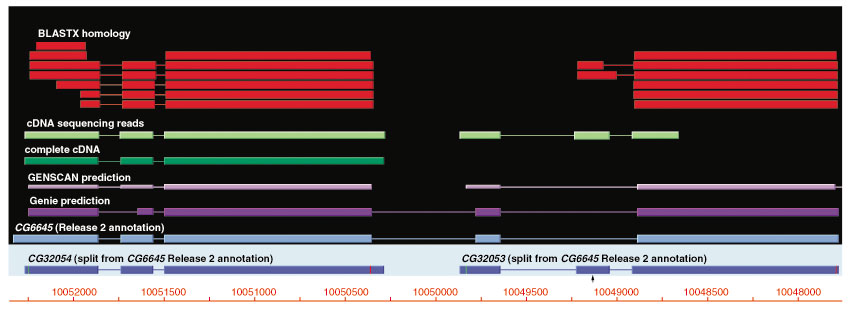
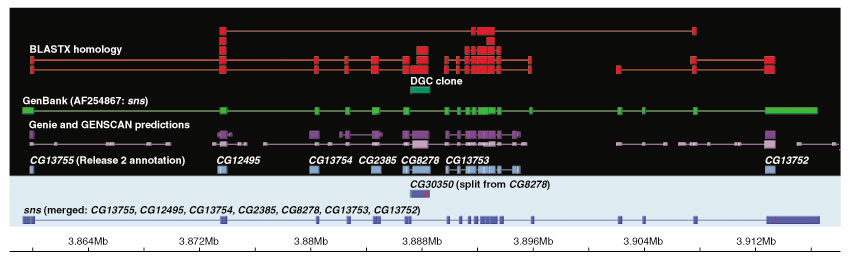
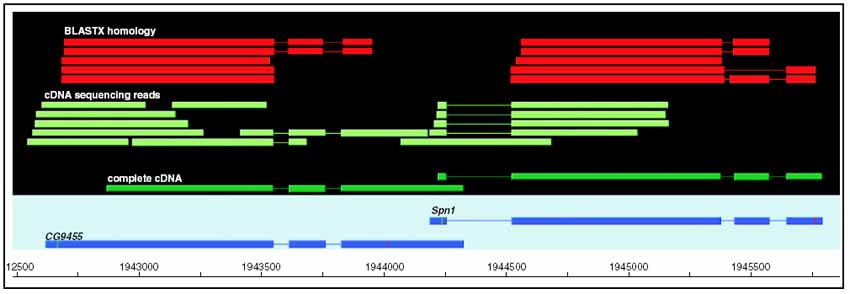
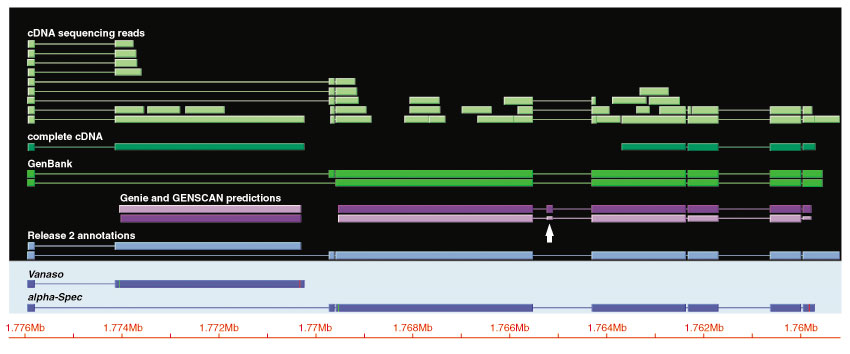
Although the number of predicted protein-coding genes in Drosophila remains essentially unchanged, the revised annotation significantly improves gene models, resulting in structural changes to 85% of the transcripts and 45% of the predicted proteins. We annotated transposable elements and non-protein-coding RNAs as new features, and extended the annotation of untranslated (UTR) sequences and alternative transcripts to include more than 70% and 20% of genes, respectively. Finally, cDNA sequence provided evidence for dicistronic transcripts, neighboring genes with overlapping UTRs on the same DNA sequence strand, alternatively spliced genes that encode distinct, non-overlapping peptides, and numerous nested genes.
Identification of so many unusual gene models not only suggests that some mechanisms for gene regulation are more prevalent than previously believed, but also underscores the complex challenges of eukaryotic gene prediction. At present, experimental data and human curation remain essential to generate high-quality genome annotations.
黑腹果蝇基因组序列近期高质量完成,且有大量扩充的果蝇cDNA序列可供使用,这些序列与78%的预测常染色体基因匹配,这使FlyBase有机会显著改进基因组注释。我们通过直观检查每个基因、利用一套全面的编辑规则、要求每个基因模型有可追溯的证据,并将每个预测的肽段与SWISS-PROT和TrEMBL序列进行比较,使注释过程更加严格。
虽然果蝇中预测的蛋白质编码基因数量基本保持不变,但修订后的注释显著改进了基因模型,导致85%的转录本和45%的预测蛋白质发生结构变化。我们将转座元件和非蛋白质编码RNA注释为新特征,并扩展了非翻译(UTR)序列和可变转录本的注释,分别涵盖了70%以上和20%以上的基因。最后,cDNA序列为双顺反子转录本、同一DNA序列链上具有重叠UTR的相邻基因、编码不同的非重叠肽段的可变剪接基因以及众多嵌套基因提供了证据。
识别出如此多不寻常的基因模型不仅表明某些基因调控机制比以前认为的更为普遍,也凸显了真核基因预测的复杂挑战。目前,实验数据和人工编辑对于生成高质量的基因组注释仍然至关重要。