State Key Laboratory for Diagnosis and Treatment of Infectious Diseases, the First Affiliated Hospital, College of Medicine, Zhejiang University, Hangzhou, Zhejiang 310003, China.
BMC Genomics. 2010 Sep 7;11:488. doi: 10.1186/1471-2164-11-488.
Bacterial vaginosis (BV) is an ecological disorder of the vaginal microbiota that affects millions of women annually, and is associated with numerous adverse health outcomes including pre-term birth and the acquisition of sexually transmitted infections. However, little is known about the overall structure and composition of vaginal microbial communities; most of the earlier studies focused on predominant vaginal bacteria in the process of BV. In the present study, the diversity and richness of vaginal microbiota in 50 BV positive and 50 healthy women from China were investigated using culture-independent PCR-denaturing gradient gel electrophoresis (DGGE) and barcoded 454 pyrosequencing methods, and validated by quantitative PCR.
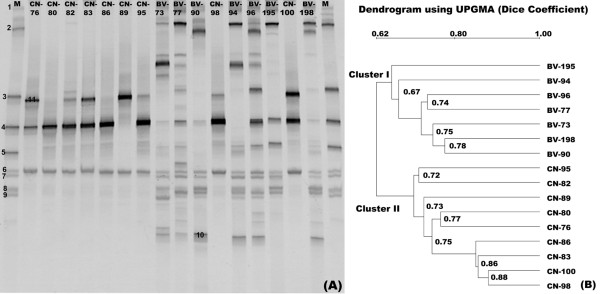
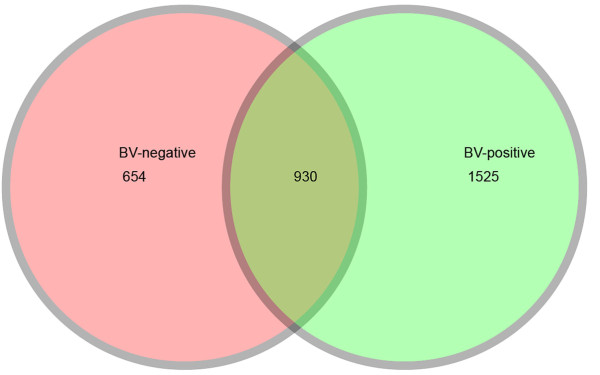
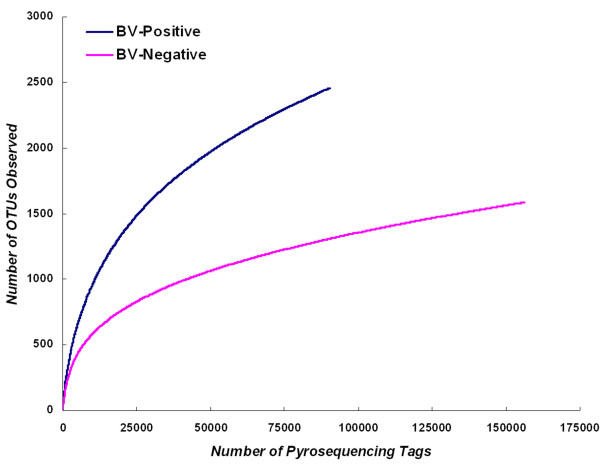
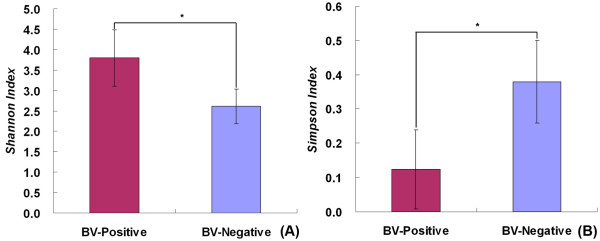
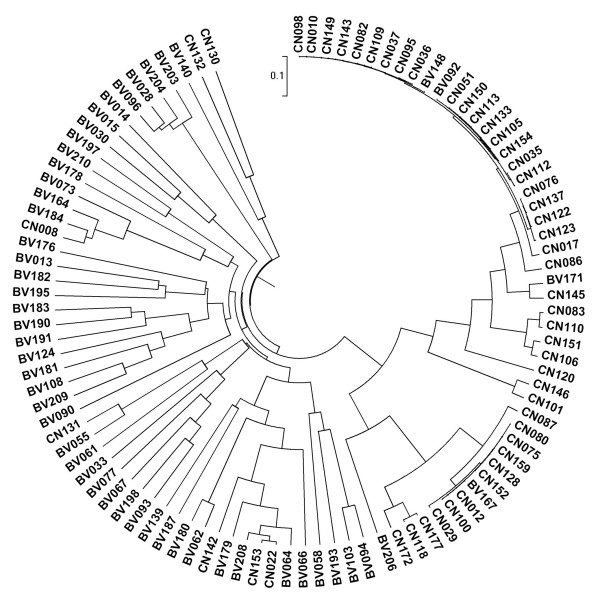
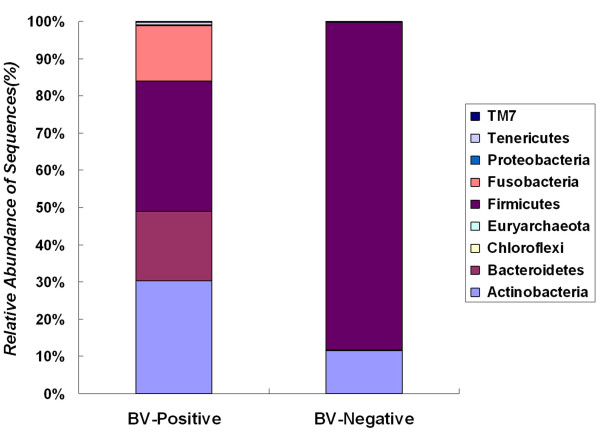
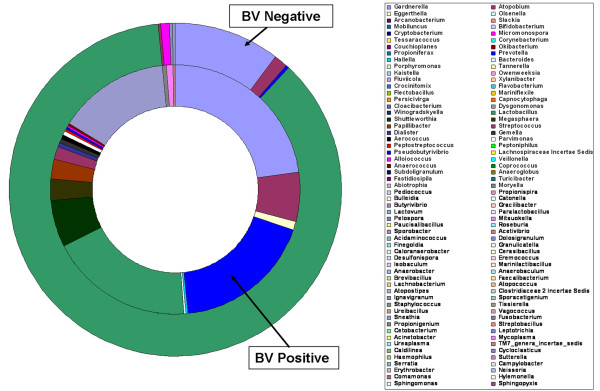
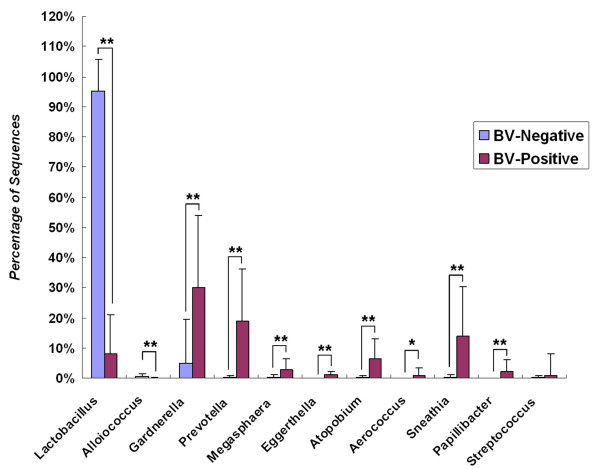
Our data demonstrated that there was a profound shift in the absolute and relative abundances of bacterial species present in the vagina when comparing populations associated with healthy and diseased conditions. In spite of significant interpersonal variations, the diversity of vaginal microbiota in the two groups could be clearly divided into two clusters. A total of 246,359 high quality pyrosequencing reads was obtained for evaluating bacterial diversity and 24,298 unique sequences represented all phylotypes. The most predominant phyla of bacteria identified in the vagina belonged to Firmicutes, Bacteroidetes, Actinobacteria and Fusobacteria. The higher number of phylotypes in BV positive women over healthy is consistent with the results of previous studies and a large number of low-abundance taxa which were missed in previous studies were revealed. Although no single bacterium could be identified as a specific marker for healthy over diseased conditions, three phyla - Bacteroidetes, Actinobacteria and Fusobacteria, and eight genera including Gardnerella, Atopobium, Megasphaera, Eggerthella, Aerococcus, Leptotrichia/Sneathia, Prevotella and Papillibacter were strongly associated with BV (p < 0.05). These genera are potentially excellent markers and could be used as targets for clinical BV diagnosis by molecular approaches.
The data presented here have clearly profiled the overall structure of vaginal communities and clearly demonstrated that BV is associated with a dramatic increase in the taxonomic richness and diversity of vaginal microbiota. The study also provides the most comprehensive picture of the vaginal community structure and the bacterial ecosystem, and significantly contributes to the current understanding of the etiology of BV.
细菌性阴道病(BV)是一种阴道微生物群落的生态失调,每年影响数百万妇女,与许多不良健康后果有关,包括早产和获得性传播感染。然而,人们对阴道微生物群落的整体结构和组成知之甚少;早期的大多数研究都集中在 BV 过程中主要的阴道细菌上。本研究采用非依赖性 PCR-变性梯度凝胶电泳(DGGE)和条形码 454 焦磷酸测序方法,对来自中国的 50 例 BV 阳性和 50 例健康女性的阴道微生物群落的多样性和丰富度进行了研究,并通过定量 PCR 进行了验证。
我们的数据表明,比较健康和患病人群时,阴道中存在的细菌种类的绝对和相对丰度发生了深刻变化。尽管存在显著的人际差异,但两组阴道微生物群落的多样性可以清楚地分为两个聚类。共获得 246359 个高质量的焦磷酸测序读数来评估细菌多样性,24298 个独特序列代表了所有的类群。阴道中最主要的细菌门属于厚壁菌门、拟杆菌门、放线菌门和梭杆菌门。BV 阳性女性的类群数量高于健康女性,这与之前的研究结果一致,并揭示了大量之前研究中错过的低丰度类群。尽管没有一种细菌可以被确定为健康与患病状态的特定标志物,但有三个门——拟杆菌门、放线菌门和梭杆菌门,以及八个属,包括加德纳菌属、阿托波菌属、巨球形菌属、 Eggerthella 属、Aerococcus 属、Leptotrichia/Sneathia 属、普雷沃氏菌属和乳杆菌属,与 BV 密切相关(p < 0.05)。这些属可能是很好的标志物,可以作为通过分子方法进行临床 BV 诊断的靶点。
本研究清楚地描绘了阴道群落的整体结构,并清楚地表明,BV 与阴道微生物群落的分类丰富度和多样性显著增加有关。该研究还提供了阴道群落结构和细菌生态系统的最全面的图片,并大大有助于当前对 BV 病因的理解。