Department of Physiology and Integrated Biosystems, College of Medicine, Inje University, Busan 614-735, South Korea.
BMC Bioinformatics. 2010 Sep 17;11:467. doi: 10.1186/1471-2105-11-467.
There is an increasing demand to assemble and align large-scale biological sequence data sets. The commonly used multiple sequence alignment programs are still limited in their ability to handle very large amounts of sequences because the system lacks a scalable high-performance computing (HPC) environment with a greatly extended data storage capacity.
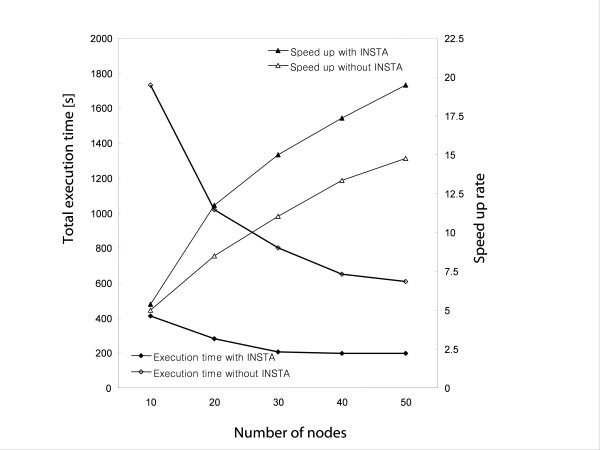
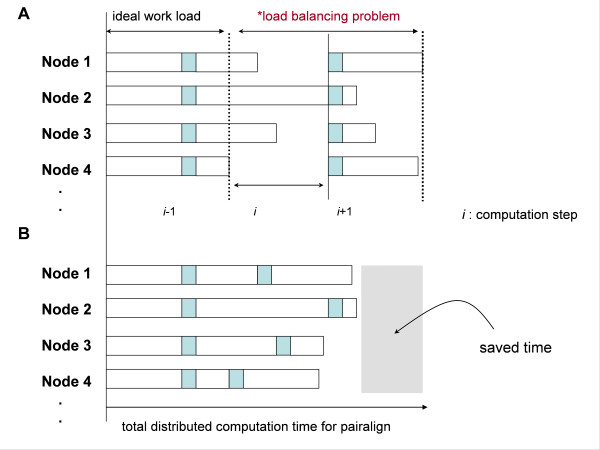
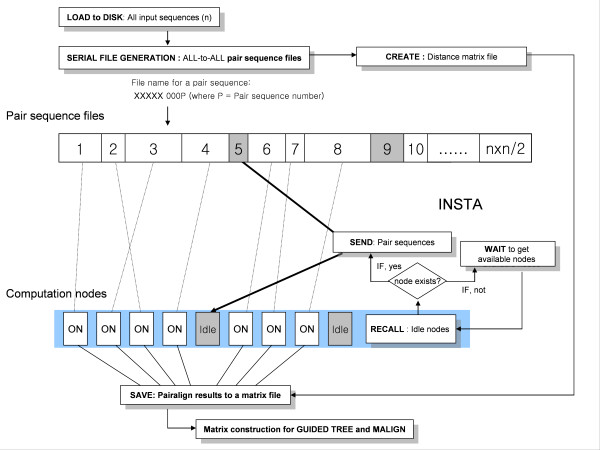
We designed ClustalXeed, a software system for multiple sequence alignment with incremental improvements over previous versions of the ClustalX and ClustalW-MPI software. The primary advantage of ClustalXeed over other multiple sequence alignment software is its ability to align a large family of protein or nucleic acid sequences. To solve the conventional memory-dependency problem, ClustalXeed uses both physical random access memory (RAM) and a distributed file-allocation system for distance matrix construction and pair-align computation. The computation efficiency of disk-storage system was markedly improved by implementing an efficient load-balancing algorithm, called "idle node-seeking task algorithm" (INSTA). The new editing option and the graphical user interface (GUI) provide ready access to a parallel-computing environment for users who seek fast and easy alignment of large DNA and protein sequence sets.
ClustalXeed can now compute a large volume of biological sequence data sets, which were not tractable in any other parallel or single MSA program. The main developments include: 1) the ability to tackle larger sequence alignment problems than possible with previous systems through markedly improved storage-handling capabilities. 2) Implementing an efficient task load-balancing algorithm, INSTA, which improves overall processing times for multiple sequence alignment with input sequences of non-uniform length. 3) Support for both single PC and distributed cluster systems.
人们对于组装和对齐大规模生物序列数据集的需求日益增长。目前常用的多序列比对程序仍然受到其处理大量序列能力的限制,因为系统缺乏可扩展的高性能计算(HPC)环境,其数据存储容量也非常有限。
我们设计了 ClustalXeed,这是一种具有增量改进的多序列比对软件系统,与 ClustalX 和 ClustalW-MPI 软件的先前版本相比。ClustalXeed 相对于其他多序列比对软件的主要优势在于其能够对齐一大类蛋白质或核酸序列。为了解决传统的内存依赖性问题,ClustalXeed 同时使用物理随机访问内存(RAM)和分布式文件分配系统来构建距离矩阵和对排列计算。通过实现高效的负载平衡算法,称为“空闲节点搜索任务算法”(INSTA),磁盘存储系统的计算效率得到了显著提高。新的编辑选项和图形用户界面(GUI)为寻求快速、轻松对齐大型 DNA 和蛋白质序列集的用户提供了对并行计算环境的便捷访问。
ClustalXeed 现在可以计算大量的生物序列数据集,这在任何其他并行或单一 MSA 程序中都是不可行的。主要的发展包括:1)通过显著提高存储处理能力,能够处理比以前的系统更大的序列比对问题。2)实现了高效的任务负载平衡算法 INSTA,它提高了具有非均匀长度输入序列的多序列比对的整体处理时间。3)支持单台 PC 和分布式集群系统。