Program in Computational Biology and Bioinformatics, Yale University, New Haven, Connecticut, United States of America.
PLoS Comput Biol. 2011 Jan 6;7(1):e1001050. doi: 10.1371/journal.pcbi.1001050.
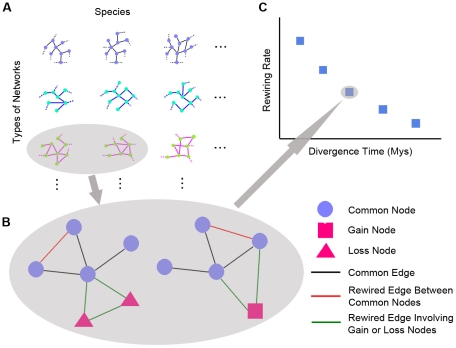
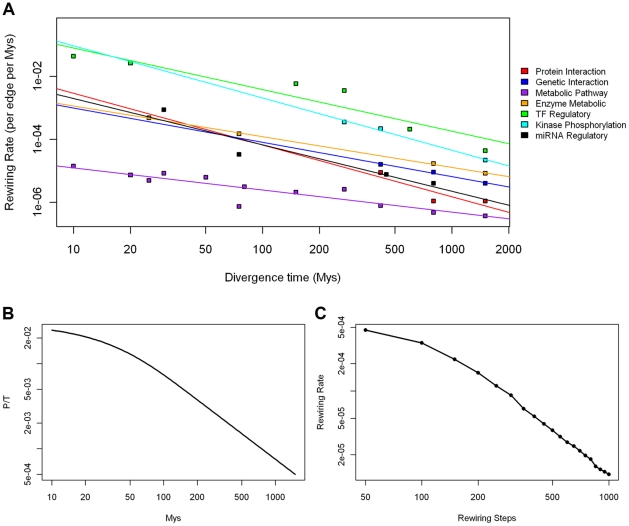
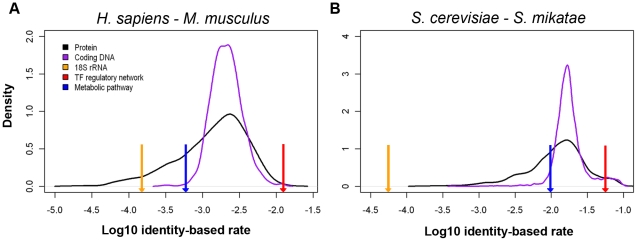
We have accumulated a large amount of biological network data and expect even more to come. Soon, we anticipate being able to compare many different biological networks as we commonly do for molecular sequences. It has long been believed that many of these networks change, or "rewire", at different rates. It is therefore important to develop a framework to quantify the differences between networks in a unified fashion. We developed such a formalism based on analogy to simple models of sequence evolution, and used it to conduct a systematic study of network rewiring on all the currently available biological networks. We found that, similar to sequences, biological networks show a decreased rate of change at large time divergences, because of saturation in potential substitutions. However, different types of biological networks consistently rewire at different rates. Using comparative genomics and proteomics data, we found a consistent ordering of the rewiring rates: transcription regulatory, phosphorylation regulatory, genetic interaction, miRNA regulatory, protein interaction, and metabolic pathway network, from fast to slow. This ordering was found in all comparisons we did of matched networks between organisms. To gain further intuition on network rewiring, we compared our observed rewirings with those obtained from simulation. We also investigated how readily our formalism could be mapped to other network contexts; in particular, we showed how it could be applied to analyze changes in a range of "commonplace" networks such as family trees, co-authorships and linux-kernel function dependencies.
我们已经积累了大量的生物网络数据,预计还会有更多的数据。很快,我们预计能够像比较分子序列那样,比较许多不同的生物网络。长期以来,人们一直认为这些网络中的许多网络会以不同的速度发生变化或“重新布线”。因此,开发一种以统一方式量化网络之间差异的框架非常重要。我们基于类比简单序列进化模型开发了这样一种形式主义,并将其用于对所有现有生物网络的网络重连进行系统研究。我们发现,与序列类似,由于潜在替代物的饱和,生物网络在较大的时间分歧时变化速度降低。然而,不同类型的生物网络始终以不同的速度进行重连。使用比较基因组学和蛋白质组学数据,我们发现重连率存在一致的排序:转录调控、磷酸化调控、遗传相互作用、miRNA 调控、蛋白质相互作用和代谢途径网络,从快到慢。在我们对生物体之间匹配网络进行的所有比较中,都发现了这种排序。为了进一步深入了解网络重连,我们将观察到的重连与模拟得到的重连进行了比较。我们还研究了我们的形式主义可以在多大程度上映射到其他网络环境;特别是,我们展示了如何将其应用于分析一系列“常见”网络(如族谱、合著关系和 Linux 内核功能依赖关系)的变化。