Department of Pathology and Yale Cancer Center, Yale University School of Medicine, New Haven, Connecticut, USA.
BMC Genomics. 2011 May 11;12:230. doi: 10.1186/1471-2164-12-230.
Genomic aberrations can be used to determine cancer diagnosis and prognosis. Clinically relevant novel aberrations can be discovered using high-throughput assays such as Single Nucleotide Polymorphism (SNP) arrays and next-generation sequencing, which typically provide aggregate signals of many cells at once. However, heterogeneity of tumor subclones dramatically complicates the task of detecting aberrations.
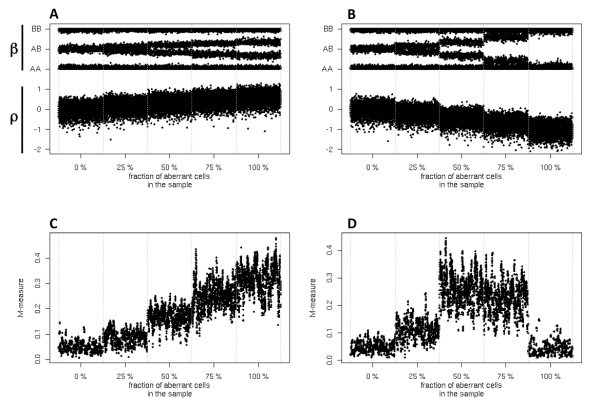
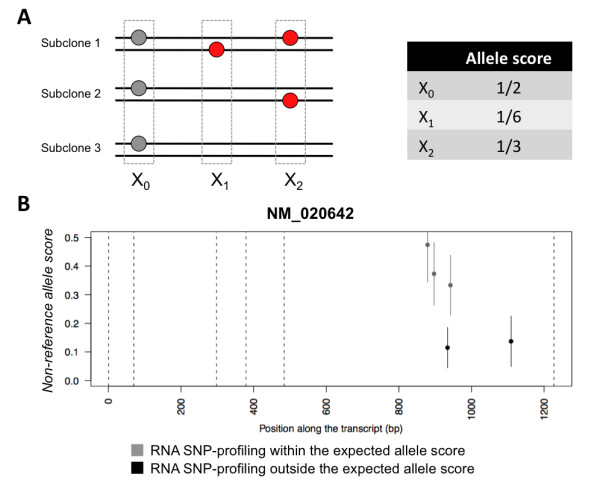
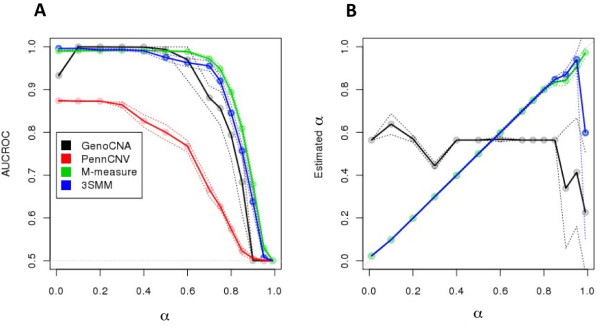
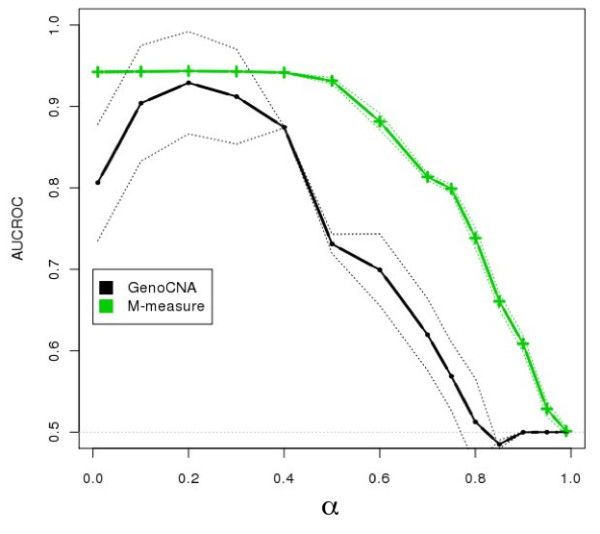
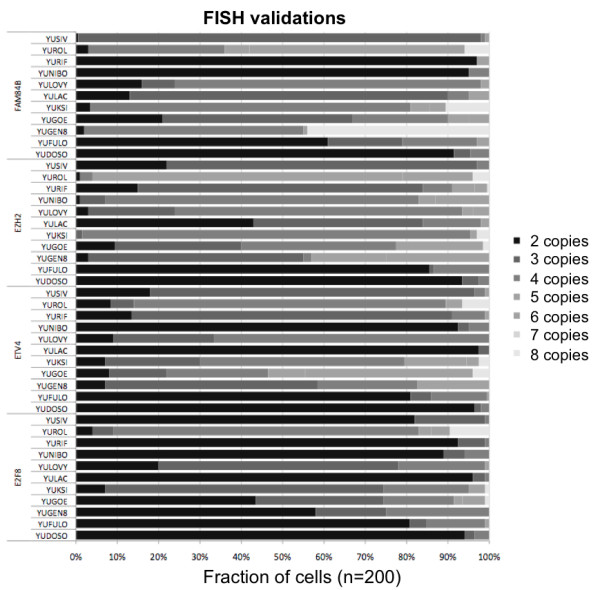
The aggregate signal of a population of subclones can be described as a linear system of equations. We employed a measure of allelic imbalance and total amount of DNA to characterize each locus by the copy number status (gain, loss or neither) of the strongest subclonal component. We designed simulated data to compare our measure to existing approaches and we analyzed SNP-arrays from 30 melanoma samples and transcriptome sequencing (RNA-Seq) from one melanoma sample.We showed that any system describing aggregate subclonal signals is underdetermined, leading to non-unique solutions for the exact copy number profile of subclones. For this reason, our illustrative measure was more robust than existing Hidden Markov Model (HMM) based tools in inferring the aberration status, as indicated by tests on simulated data. This higher robustness contributed in identifying numerous aberrations in several loci of melanoma samples. We validated the heterogeneity and aberration status within single biopsies by fluorescent in situ hybridization of four affected and transcriptionally up-regulated genes E2F8, ETV4, EZH2 and FAM84B in 11 melanoma cell lines. Heterogeneity was further demonstrated in the analysis of allelic imbalance changes along single exons from melanoma RNA-Seq.
These studies demonstrate how subclonal heterogeneity, prevalent in tumor samples, is reflected in aggregate signals measured by high-throughput techniques. Our proposed approach yields high robustness in detecting copy number alterations using high-throughput technologies and has the potential to identify specific subclonal markers from next-generation sequencing data.
基因组异常可用于确定癌症的诊断和预后。高通量检测,如单核苷酸多态性(SNP)阵列和下一代测序,可以发现具有临床意义的新型异常,这些检测方法可以一次性提供大量细胞的综合信号。然而,肿瘤亚克隆的异质性极大地增加了检测异常的难度。
亚克隆群体的综合信号可以用线性方程组来描述。我们使用等位基因失衡和 DNA 总量的度量来描述每个基因座的拷贝数状态(增益、缺失或不增益/缺失),即最强亚克隆成分的拷贝数状态。我们设计了模拟数据来比较我们的方法和现有的方法,并分析了 30 个黑色素瘤样本的 SNP 阵列和一个黑色素瘤样本的转录组测序(RNA-Seq)数据。我们表明,任何描述综合亚克隆信号的系统都是欠定的,这导致了亚克隆的精确拷贝数图谱的非唯一解。因此,我们的说明性方法在推断异常状态方面比现有的基于隐马尔可夫模型(HMM)的工具更稳健,这在模拟数据的测试中得到了体现。这种更高的稳健性有助于识别多个黑色素瘤样本中多个基因座的异常。我们通过对 11 个黑色素瘤细胞系中 4 个受影响且转录上调的基因(E2F8、ETV4、EZH2 和 FAM84B)进行荧光原位杂交,验证了单个活检样本中的异质性和异常状态。我们还通过黑色素瘤 RNA-Seq 中单外显子的等位基因失衡变化分析进一步证明了异质性。
这些研究表明,肿瘤样本中普遍存在的亚克隆异质性是如何反映在高通量技术测量的综合信号中的。我们提出的方法在使用高通量技术检测拷贝数改变方面具有很高的稳健性,并且有可能从下一代测序数据中识别特定的亚克隆标记。