McMaster Ancient DNA Centre, Department of Anthropology, McMaster University, 1280 Main Street West, Hamilton, Ontario L8S 4L8, Canada.
Nature. 2011 Oct 12;478(7370):506-10. doi: 10.1038/nature10549.
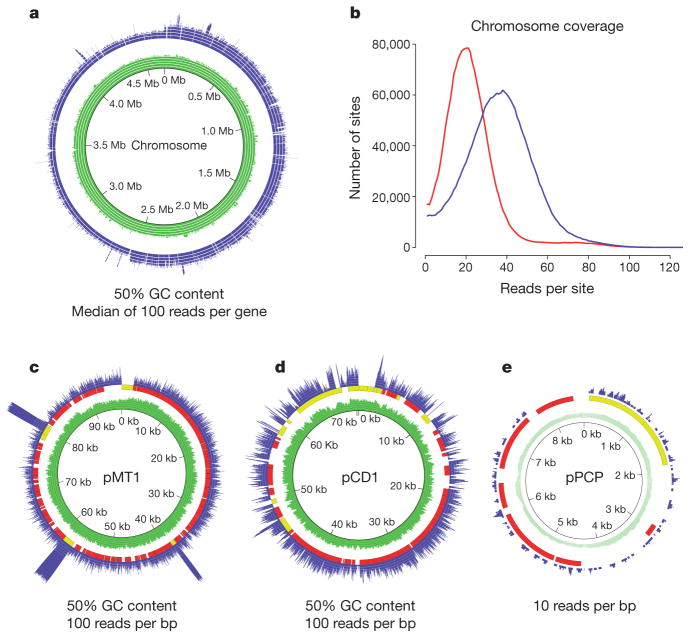
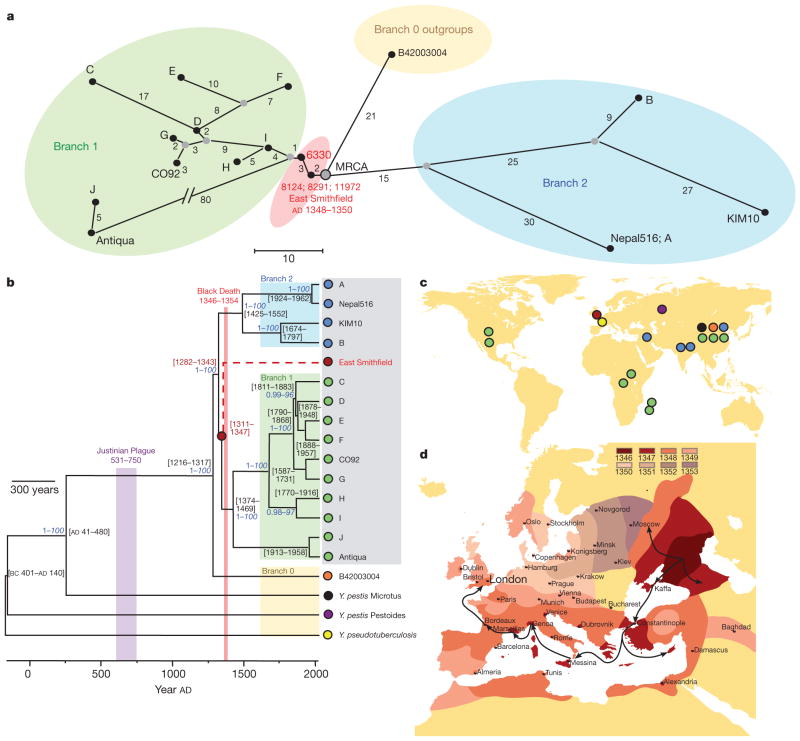
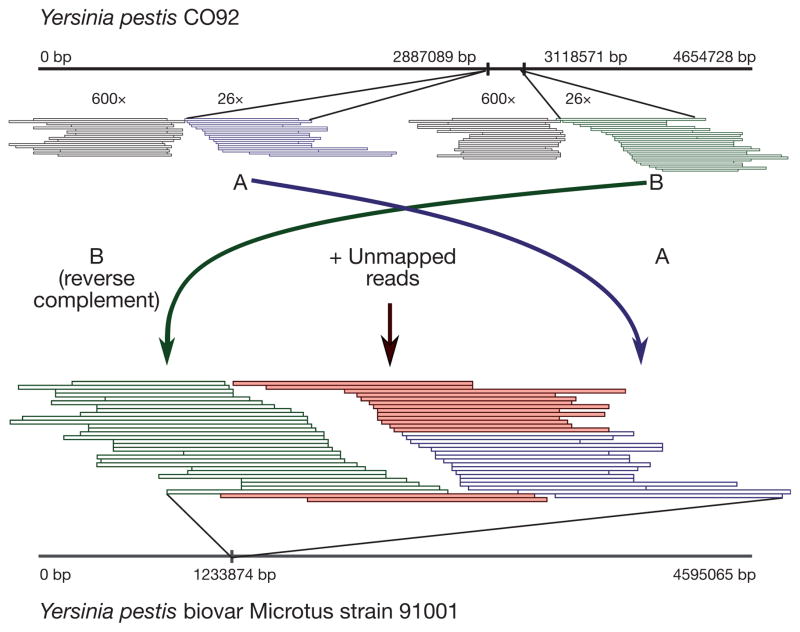
Technological advances in DNA recovery and sequencing have drastically expanded the scope of genetic analyses of ancient specimens to the extent that full genomic investigations are now feasible and are quickly becoming standard. This trend has important implications for infectious disease research because genomic data from ancient microbes may help to elucidate mechanisms of pathogen evolution and adaptation for emerging and re-emerging infections. Here we report a reconstructed ancient genome of Yersinia pestis at 30-fold average coverage from Black Death victims securely dated to episodes of pestilence-associated mortality in London, England, 1348-1350. Genetic architecture and phylogenetic analysis indicate that the ancient organism is ancestral to most extant strains and sits very close to the ancestral node of all Y. pestis commonly associated with human infection. Temporal estimates suggest that the Black Death of 1347-1351 was the main historical event responsible for the introduction and widespread dissemination of the ancestor to all currently circulating Y. pestis strains pathogenic to humans, and further indicates that contemporary Y. pestis epidemics have their origins in the medieval era. Comparisons against modern genomes reveal no unique derived positions in the medieval organism, indicating that the perceived increased virulence of the disease during the Black Death may not have been due to bacterial phenotype. These findings support the notion that factors other than microbial genetics, such as environment, vector dynamics and host susceptibility, should be at the forefront of epidemiological discussions regarding emerging Y. pestis infections.
DNA 回收和测序技术的进步极大地扩展了对古代标本进行遗传分析的范围,以至于现在可以进行全基因组研究,并且该研究正在迅速成为标准。这一趋势对传染病研究具有重要意义,因为来自古代微生物的基因组数据可能有助于阐明病原体进化和适应新发和再发感染的机制。在这里,我们报道了从安全定年为 1348-1350 年英格兰伦敦瘟疫相关死亡事件的黑死病受害者中获得的 30 倍平均覆盖率的鼠疫耶尔森氏菌重建古代基因组。遗传结构和系统发育分析表明,该古代生物是大多数现存菌株的祖先,并且非常接近与人类感染相关的所有鼠疫耶尔森氏菌的祖先节点。时间估计表明,1347-1351 年的黑死病是导致所有当前流行的人类致病鼠疫耶尔森氏菌菌株的祖先传入和广泛传播的主要历史事件,并进一步表明当代鼠疫耶尔森氏菌流行起源于中世纪。与现代基因组的比较没有发现中世纪生物中独特的衍生位置,这表明在黑死病期间疾病的感知毒力增加可能不是由于细菌表型。这些发现支持这样一种观点,即除了微生物遗传学之外的其他因素,例如环境、媒介动力学和宿主易感性,应该成为关于新发鼠疫耶尔森氏菌感染的流行病学讨论的前沿。