Altwasser Robert, Linde Jörg, Buyko Ekaterina, Hahn Udo, Guthke Reinhard
Research Group Systems Biology/Bioinformatics, Leibniz Institute for Natural Product Research and Infection Biology - Hans Knoell Institute Jena, Germany.
Front Microbiol. 2012 Feb 16;3:51. doi: 10.3389/fmicb.2012.00051. eCollection 2012.
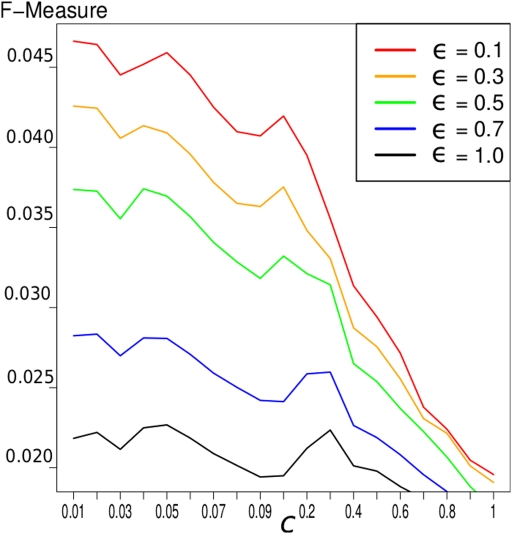
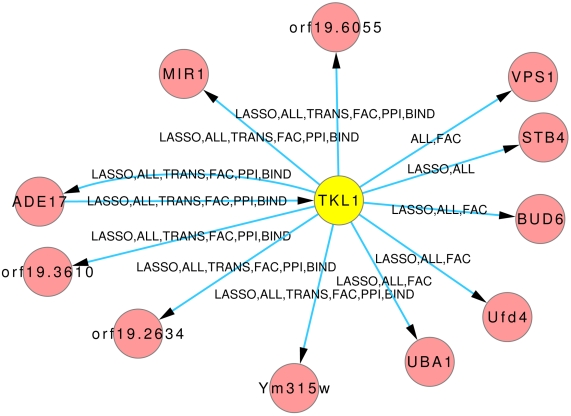
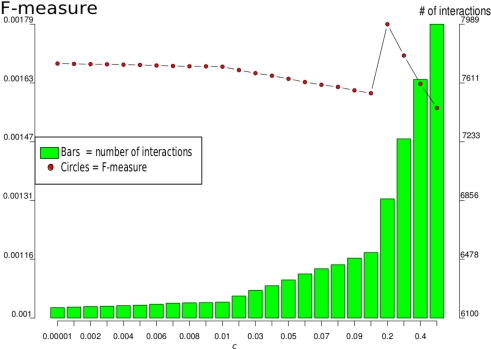
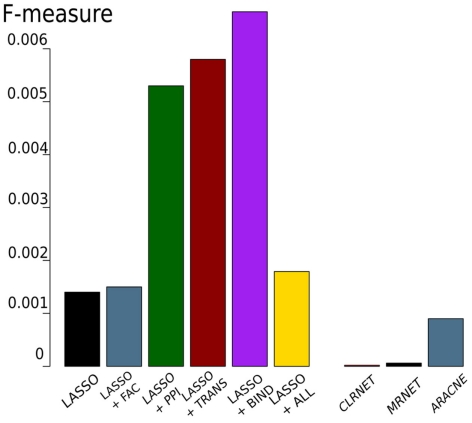
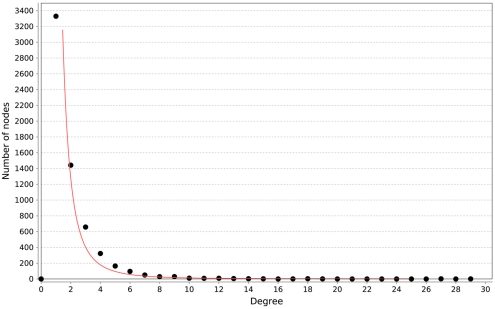
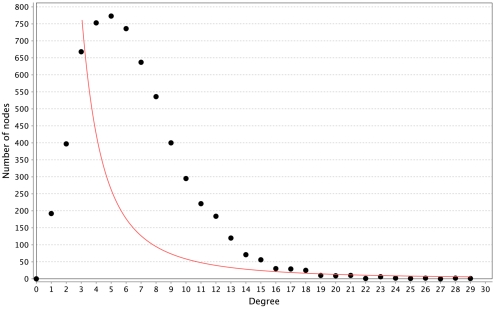
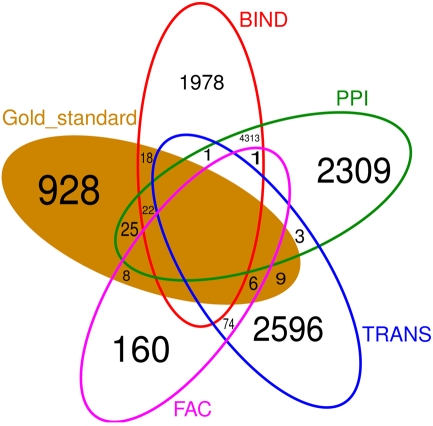
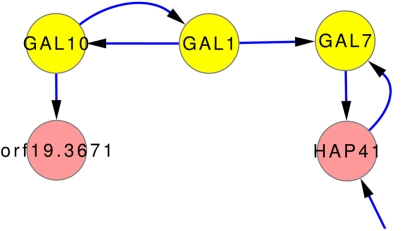
Discovery of essential genes in pathogenic organisms is an important step in the development of new medication. Despite a growing number of genome data available, little is known about C. albicans, a major fungal pathogen. Most of the human population carries C. albicans as commensal, but it can cause systemic infection that may lead to the death of the host if the immune system has deteriorated. In many organisms central nodes in the interaction network (hubs) play a crucial role for information and energy transport. Knock-outs of such hubs often lead to lethal phenotypes making them interesting drug targets. To identify these central genes via topological analysis, we inferred gene regulatory networks that are sparse and scale-free. We collected information from various sources to complement the limited expression data available. We utilized a linear regression algorithm to infer genome-wide gene regulatory interaction networks. To evaluate the predictive power of our approach, we used an automated text-mining system that scanned full-text research papers for known interactions. With the help of the compendium of known interactions, we also optimize the influence of the prior knowledge and the sparseness of the model to achieve the best results. We compare the results of our approach with those of other state-of-the-art network inference methods and show that we outperform those methods. Finally we identify a number of hubs in the genome of the fungus and investigate their biological relevance.
发现致病生物中的必需基因是开发新药的重要一步。尽管可用的基因组数据越来越多,但对于主要真菌病原体白色念珠菌,人们了解甚少。大多数人携带白色念珠菌作为共生菌,但如果免疫系统恶化,它可能会引发全身感染,导致宿主死亡。在许多生物中,相互作用网络中的中心节点(枢纽)在信息和能量传输中起着关键作用。敲除这些枢纽通常会导致致死表型,使其成为有趣的药物靶点。为了通过拓扑分析识别这些中心基因,我们推断出稀疏且无标度的基因调控网络。我们从各种来源收集信息,以补充有限的可用表达数据。我们利用线性回归算法推断全基因组的基因调控相互作用网络。为了评估我们方法的预测能力,我们使用了一个自动文本挖掘系统,该系统扫描全文研究论文以寻找已知的相互作用。借助已知相互作用的汇编,我们还优化了先验知识的影响和模型的稀疏性,以获得最佳结果。我们将我们方法的结果与其他最先进的网络推断方法的结果进行比较,结果表明我们的方法优于那些方法。最后,我们在真菌基因组中识别出一些枢纽,并研究它们的生物学相关性。