Department of Zoology, University of Oxford, Oxford OX1 3PS, UK.
The Wellcome Trust Sanger Institute, Wellcome Trust Genome Campus, Hinxton CB10 1SA, UK.
Microbiology (Reading). 2012 Jun;158(Pt 6):1570-1580. doi: 10.1099/mic.0.056077-0. Epub 2012 Mar 15.
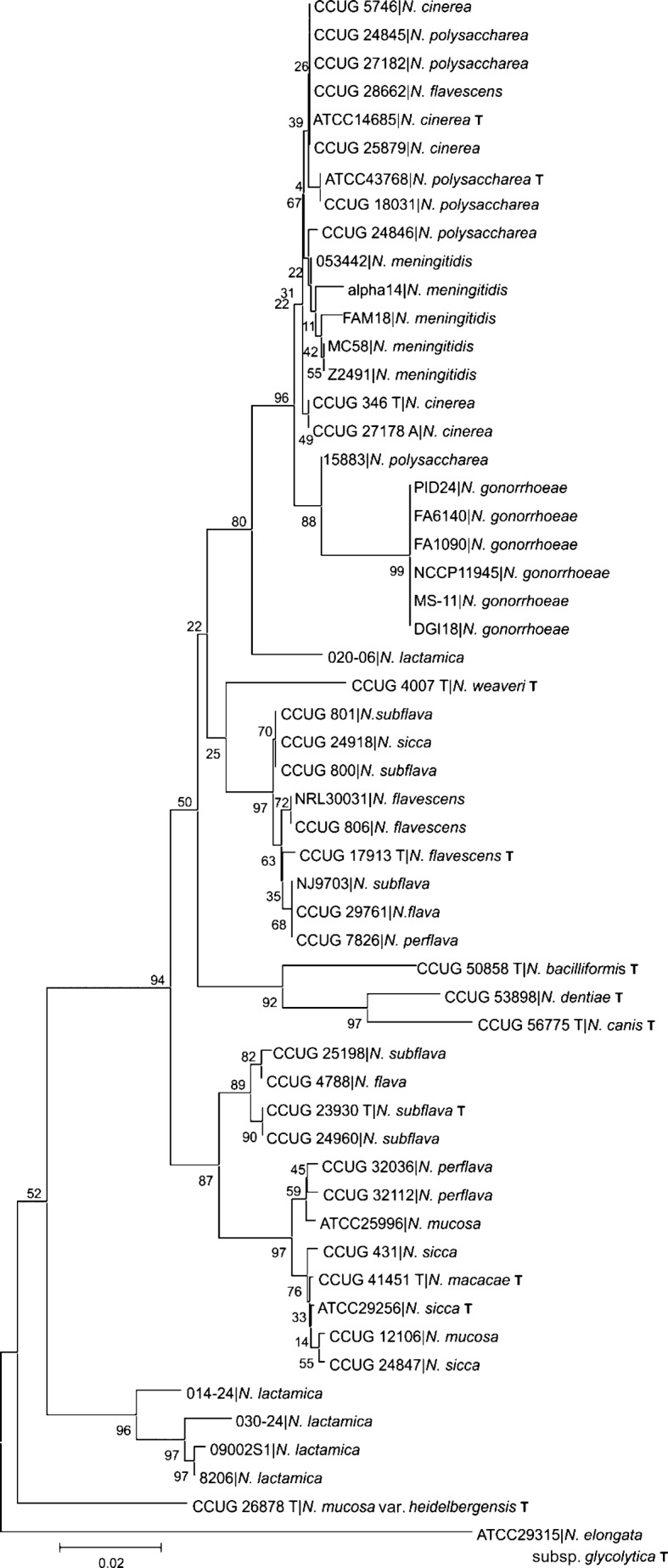
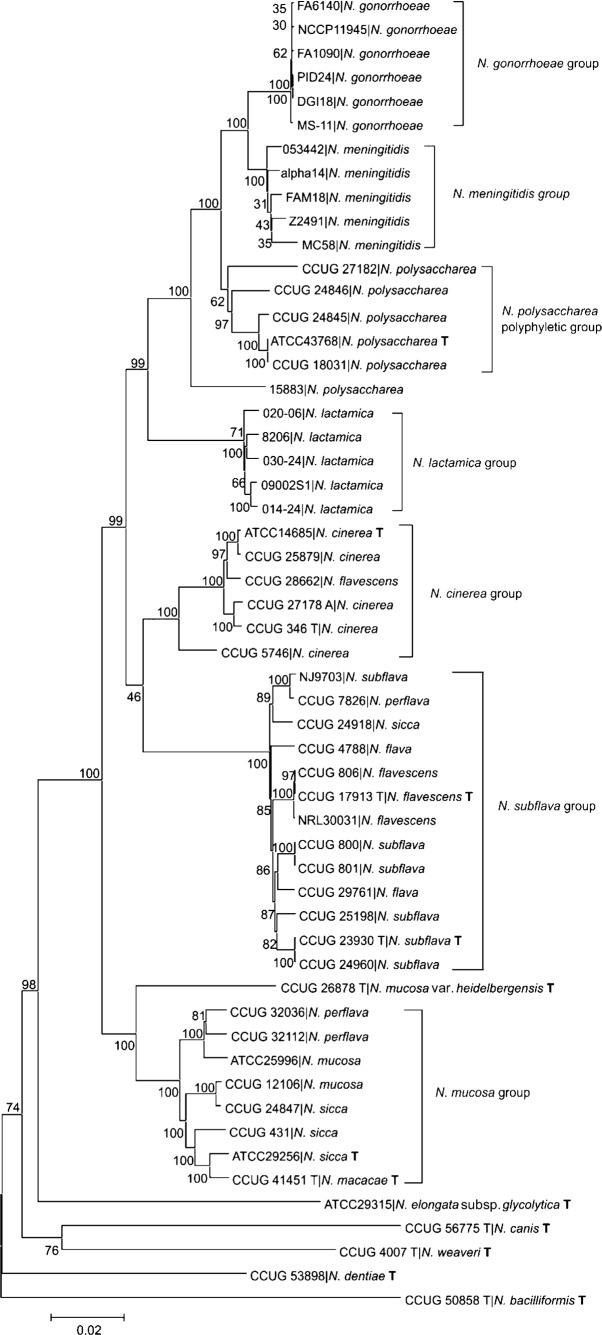
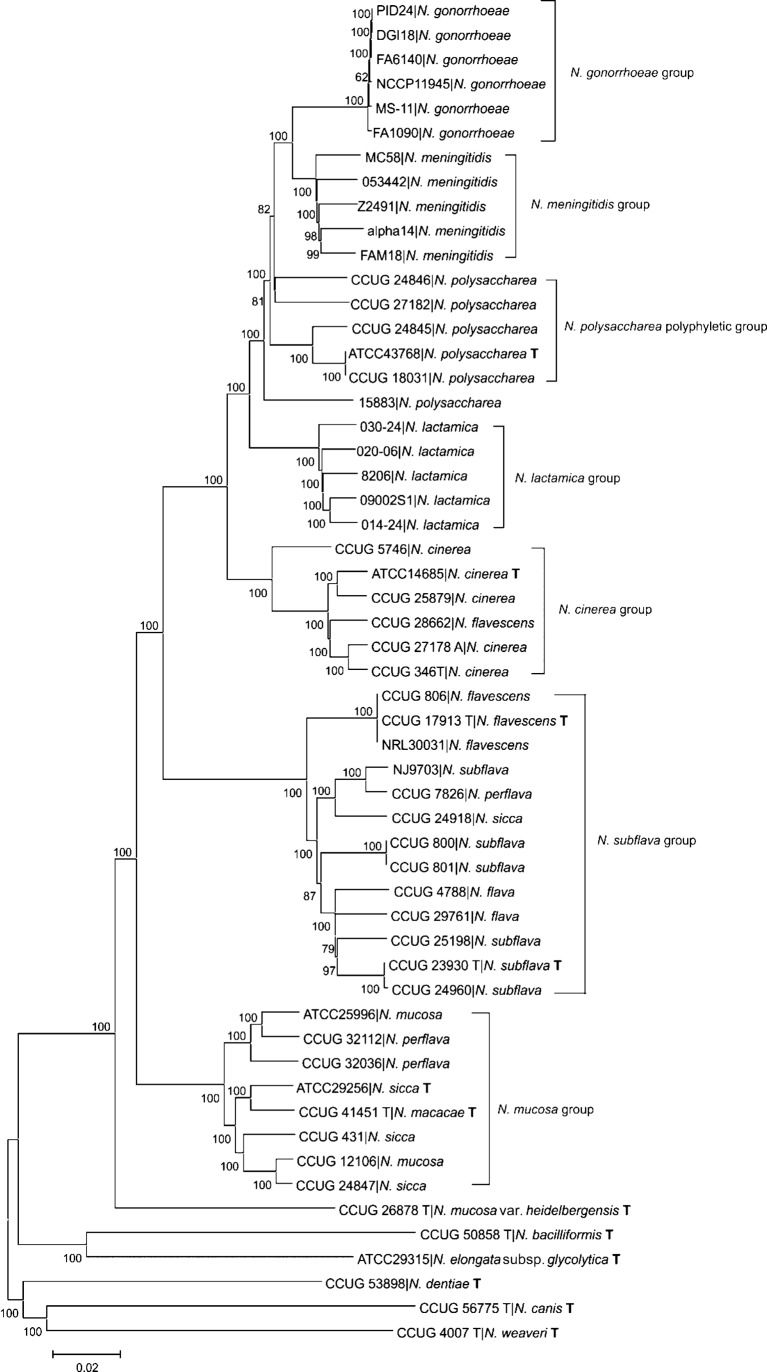
In common with other bacterial taxa, members of the genus Neisseria are classified using a range of phenotypic and biochemical approaches, which are not entirely satisfactory in assigning isolates to species groups. Recently, there has been increasing interest in using nucleotide sequences for bacterial typing and taxonomy, but to date, no broadly accepted alternative to conventional methods is available. Here, the taxonomic relationships of 55 representative members of the genus Neisseria have been analysed using whole-genome sequence data. As genetic material belonging to the accessory genome is widely shared among different taxa but not present in all isolates, this analysis indexed nucleotide sequence variation within sets of genes, specifically protein-coding genes that were present and directly comparable in all isolates. Variation in these genes identified seven species groups, which were robust to the choice of genes and phylogenetic clustering methods used. The groupings were largely, but not completely, congruent with current species designations, with some minor changes in nomenclature and the reassignment of a few isolates necessary. In particular, these data showed that isolates classified as Neisseria polysaccharea are polyphyletic and probably include more than one taxonomically distinct organism. The seven groups could be reliably and rapidly generated with sequence variation within the 53 ribosomal protein subunit (rps) genes, further demonstrating that ribosomal multilocus sequence typing (rMLST) is a practicable and powerful means of characterizing bacteria at all levels, from domain to strain.
与其他细菌分类群一样,奈瑟氏菌属的成员使用一系列表型和生化方法进行分类,这些方法在将分离物分配到物种组时并不完全令人满意。最近,人们越来越感兴趣地使用核苷酸序列进行细菌分型和分类学,但迄今为止,还没有一种广泛接受的替代传统方法。在这里,使用全基因组序列数据分析了 55 个代表性奈瑟氏菌属成员的分类关系。由于属于辅助基因组的遗传物质在不同的分类群中广泛共享,但并非所有分离物都存在,因此这种分析索引了一组基因(特别是在所有分离物中存在且可直接比较的蛋白质编码基因)内的核苷酸序列变异。这些基因的变异确定了七个物种组,这些组在选择基因和使用的系统发育聚类方法方面具有稳健性。分组在很大程度上,但并非完全与当前的物种命名一致,需要进行一些小的命名法更改和一些分离物的重新分配。特别是,这些数据表明,被分类为 Neisseria polysaccharea 的分离物是多系的,可能包含一种以上在分类上不同的生物体。通过 53 个核糖体蛋白亚基(rps)基因内的序列变异,可以可靠且快速地生成这七个组,进一步证明核糖体多位点序列分型(rMLST)是一种可行且强大的方法,可以在从域到菌株的所有水平上对细菌进行特征描述。