Gu Zuguang, Liu Jialin, Cao Kunming, Zhang Junfeng, Wang Jin
The State Key Laboratory of Pharmaceutical Biotechnology and Jiangsu Engineering Research Center for MicroRNA Biology and Biotechnology, School of Life Science, Nanjing University, Nanjing, 210093, China.
BMC Syst Biol. 2012 Jun 6;6:56. doi: 10.1186/1752-0509-6-56.
Biological pathways are important for understanding biological mechanisms. Thus, finding important pathways that underlie biological problems helps researchers to focus on the most relevant sets of genes. Pathways resemble networks with complicated structures, but most of the existing pathway enrichment tools ignore topological information embedded within pathways, which limits their applicability.
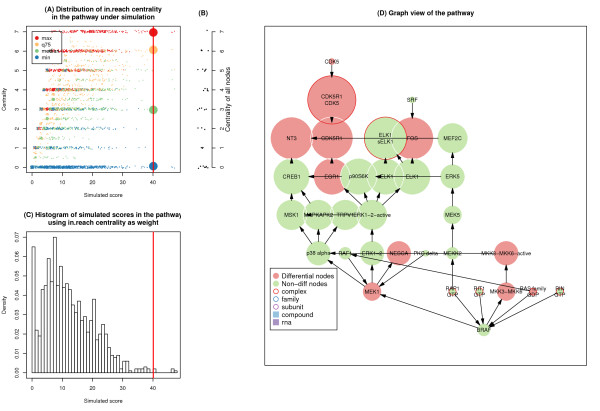
A systematic and extensible pathway enrichment method in which nodes are weighted by network centrality was proposed. We demonstrate how choice of pathway structure and centrality measurement, as well as the presence of key genes, affects pathway significance. We emphasize two improvements of our method over current methods. First, allowing for the diversity of genes' characters and the difficulty of covering gene importance from all aspects, we set centrality as an optional parameter in the model. Second, nodes rather than genes form the basic unit of pathways, such that one node can be composed of several genes and one gene may reside in different nodes. By comparing our methodology to the original enrichment method using both simulation data and real-world data, we demonstrate the efficacy of our method in finding new pathways from biological perspective.
Our method can benefit the systematic analysis of biological pathways and help to extract more meaningful information from gene expression data. The algorithm has been implemented as an R package CePa, and also a web-based version of CePa is provided.
生物途径对于理解生物学机制非常重要。因此,找到构成生物学问题基础的重要途径有助于研究人员专注于最相关的基因集。途径类似于具有复杂结构的网络,但大多数现有的途径富集工具忽略了途径中嵌入的拓扑信息,这限制了它们的适用性。
提出了一种系统且可扩展的途径富集方法,其中节点通过网络中心性进行加权。我们展示了途径结构和中心性测量的选择以及关键基因的存在如何影响途径的显著性。我们强调我们的方法相对于当前方法的两点改进。第一,考虑到基因特征的多样性以及从各个方面涵盖基因重要性的难度,我们将中心性设置为模型中的一个可选参数。第二,节点而非基因构成途径的基本单元,这样一个节点可以由几个基因组成,一个基因可能存在于不同的节点中。通过使用模拟数据和实际数据将我们的方法与原始富集方法进行比较,我们证明了我们的方法从生物学角度发现新途径的有效性。
我们的方法有助于生物途径的系统分析,并有助于从基因表达数据中提取更有意义的信息。该算法已作为一个R包CePa实现,并且还提供了基于网络的CePa版本。