Computational Biology & Biological Physics, Department of Astronomy and Theoretical Physics, Lund University, Lund, Sweden.
PLoS Comput Biol. 2013;9(8):e1003197. doi: 10.1371/journal.pcbi.1003197. Epub 2013 Aug 22.
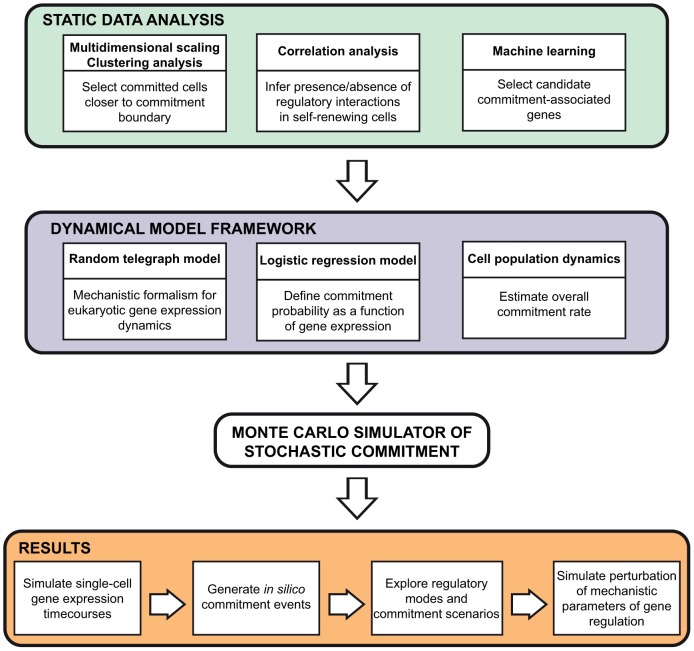
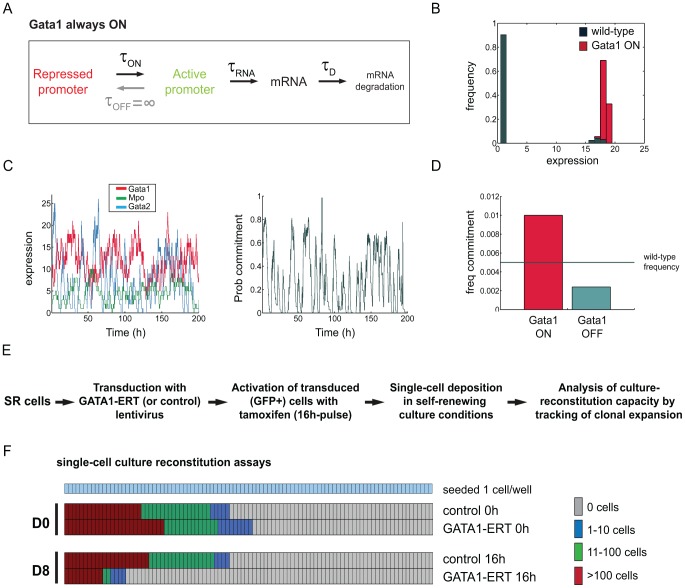
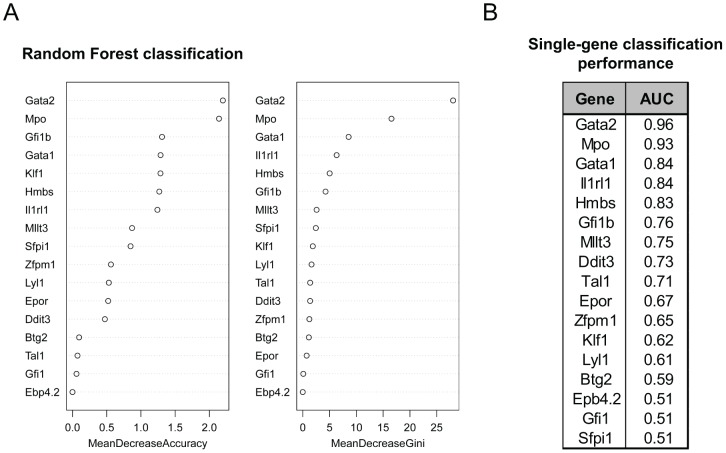
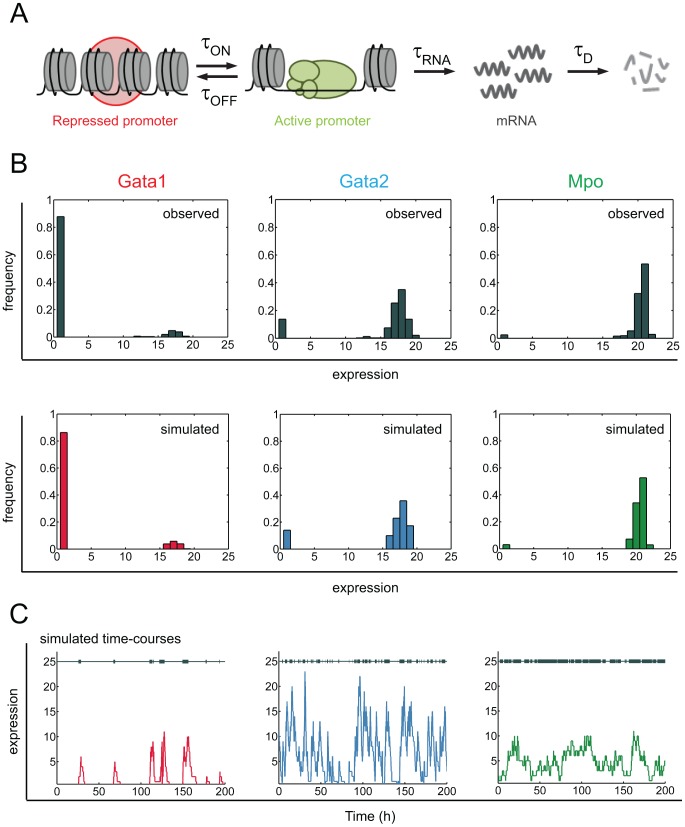
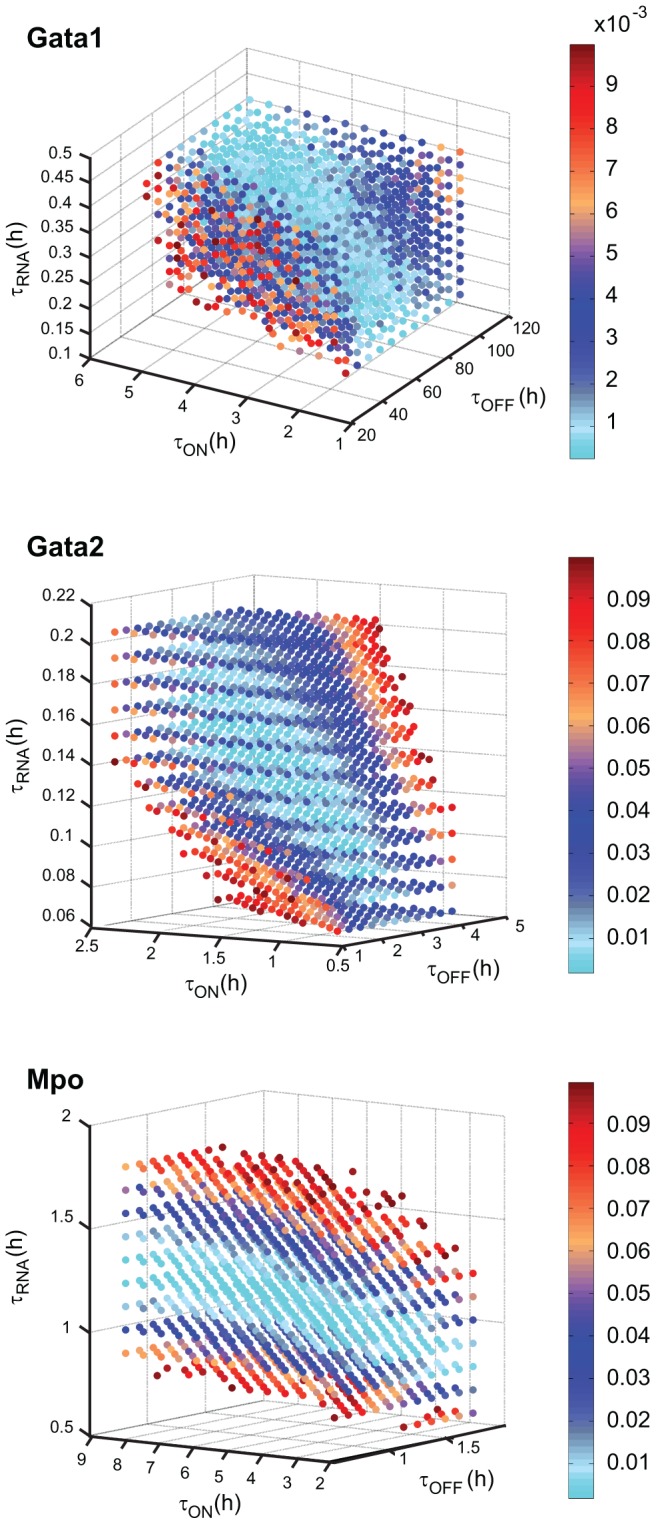
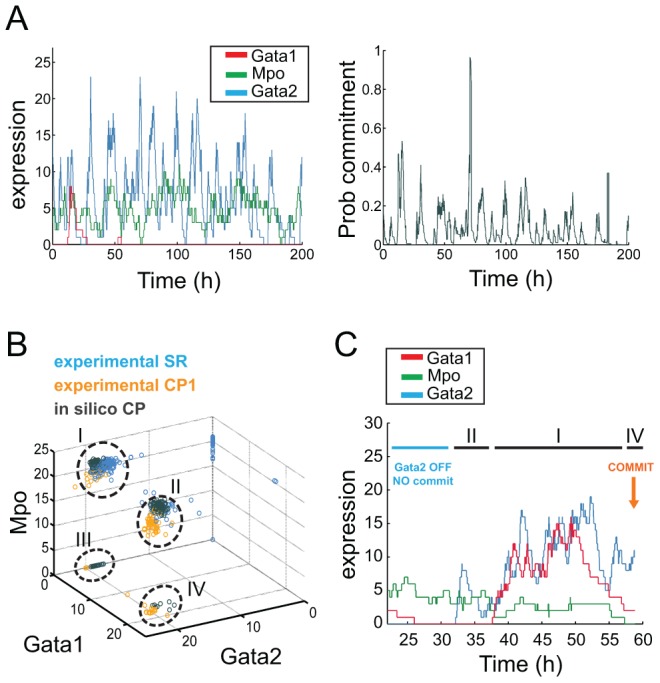
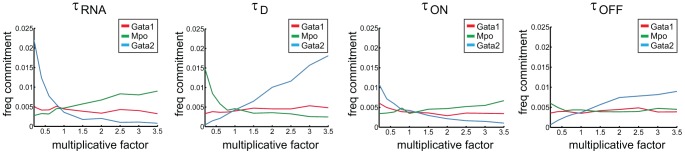
Molecular mechanisms employed by individual multipotent cells at the point of lineage commitment remain largely uncharacterized. Current paradigms span from instructive to noise-driven mechanisms. Of considerable interest is also whether commitment involves a limited set of genes or the entire transcriptional program, and to what extent gene expression configures multiple trajectories into commitment. Importantly, the transient nature of the commitment transition confounds the experimental capture of committing cells. We develop a computational framework that simulates stochastic commitment events, and affords mechanistic exploration of the fate transition. We use a combined modeling approach guided by gene expression classifier methods that infers a time-series of stochastic commitment events from experimental growth characteristics and gene expression profiling of individual hematopoietic cells captured immediately before and after commitment. We define putative regulators of commitment and probabilistic rules of transition through machine learning methods, and employ clustering and correlation analyses to interrogate gene regulatory interactions in multipotent cells. Against this background, we develop a Monte Carlo time-series stochastic model of transcription where the parameters governing promoter status, mRNA production and mRNA decay in multipotent cells are fitted to experimental static gene expression distributions. Monte Carlo time is converted to physical time using cell culture kinetic data. Probability of commitment in time is a function of gene expression as defined by a logistic regression model obtained from experimental single-cell expression data. Our approach should be applicable to similar differentiating systems where single cell data is available. Within our system, we identify robust model solutions for the multipotent population within physiologically reasonable values and explore model predictions with regard to molecular scenarios of entry into commitment. The model suggests distinct dependencies of different commitment-associated genes on mRNA dynamics and promoter activity, which globally influence the probability of lineage commitment.
个体多能细胞在谱系决定点采用的分子机制在很大程度上仍未被阐明。目前的范例涵盖了从指令性到噪声驱动的机制。同样引人关注的是,决定是否涉及有限数量的基因或整个转录程序,以及基因表达在多大程度上配置了多个进入决定的轨迹。重要的是,决定过渡的短暂性质使决定细胞的实验捕获变得复杂。我们开发了一种计算框架,可以模拟随机决定事件,并为命运转变提供机制探索。我们使用一种组合建模方法,该方法受基因表达分类器方法的指导,该方法从实验生长特征和个体造血细胞的基因表达谱推断出随机决定事件的时间序列,这些细胞在决定前后立即被捕获。我们通过机器学习方法定义了决定的假定调节剂和转换的概率规则,并采用聚类和相关分析来研究多能细胞中的基因调控相互作用。在此背景下,我们开发了一种转录的蒙特卡罗时间序列随机模型,其中控制多能细胞中启动子状态、mRNA 产生和 mRNA 衰减的参数根据实验静态基因表达分布进行拟合。使用细胞培养动力学数据将蒙特卡罗时间转换为物理时间。时间内的决定概率是由从实验单细胞表达数据获得的逻辑回归模型定义的基因表达的函数。我们的方法应该适用于具有单细胞数据的类似分化系统。在我们的系统中,我们在生理上合理的范围内为多能群体确定了稳健的模型解决方案,并根据进入决定的分子情景探索了模型预测。该模型表明不同的与决定相关的基因对 mRNA 动力学和启动子活性有不同的依赖性,这些依赖性会整体影响谱系决定的概率。