Department of Molecular and Human Genetics, CIBR Center for Computational and Integrative Biomedical Research and Program in Structural and Computational Biology & Molecular Biophysics, Baylor College of Medicine, Houston, TX 77030 and Center for Human Genetic Research, Massachusetts General Hospital, Harvard Medical School, Boston, MA 02114, USA.
Bioinformatics. 2013 Nov 1;29(21):2714-21. doi: 10.1093/bioinformatics/btt489. Epub 2013 Sep 10.
The constraints under which sequence, structure and function coevolve are not fully understood. Bringing this mutual relationship to light can reveal the molecular basis of binding, catalysis and allostery, thereby identifying function and rationally guiding protein redesign. Underlying these relationships are the epistatic interactions that occur when the consequences of a mutation to a protein are determined by the genetic background in which it occurs. Based on prior data, we hypothesize that epistatic forces operate most strongly between residues nearby in the structure, resulting in smooth evolutionary importance across the structure.
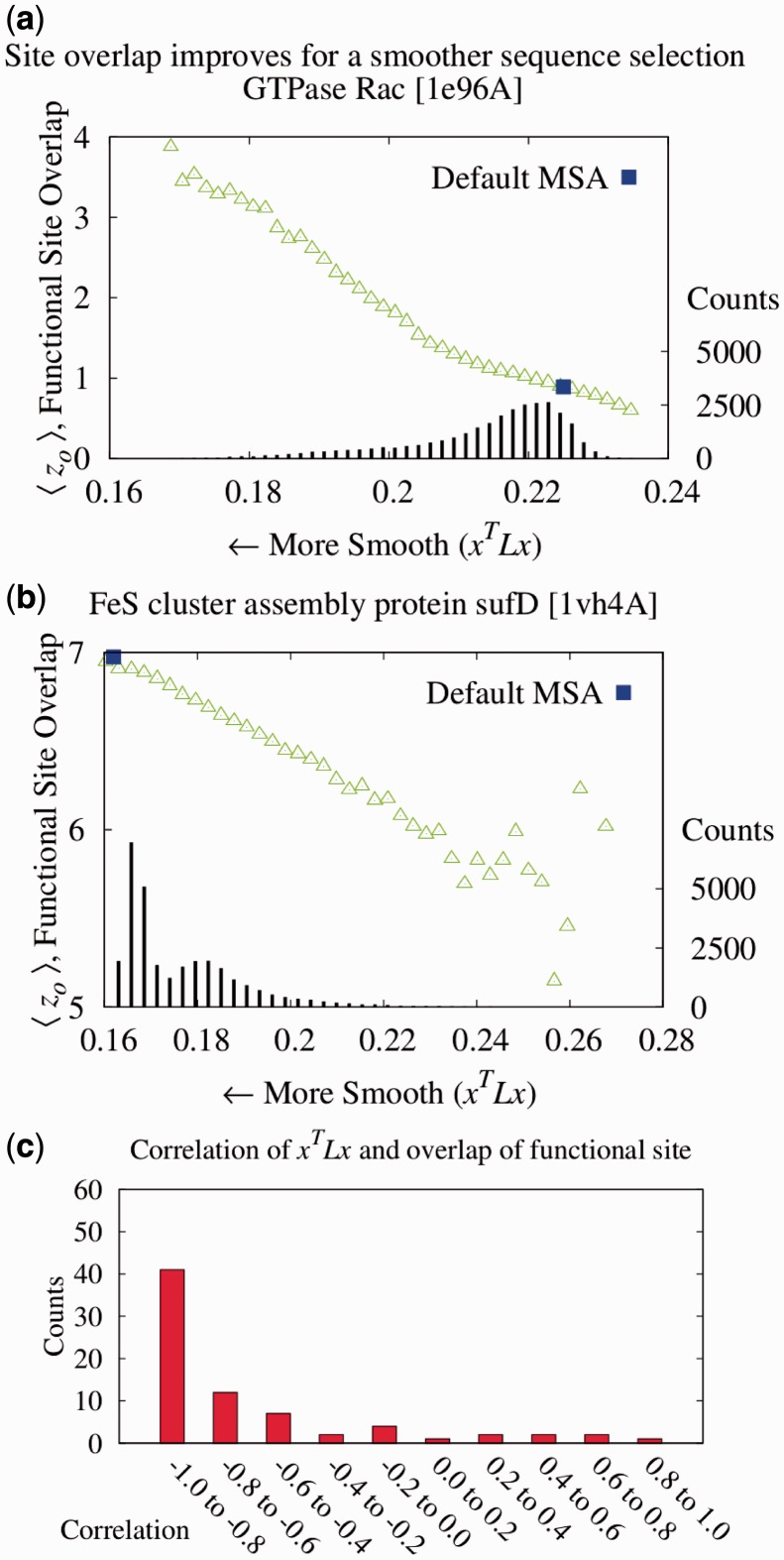
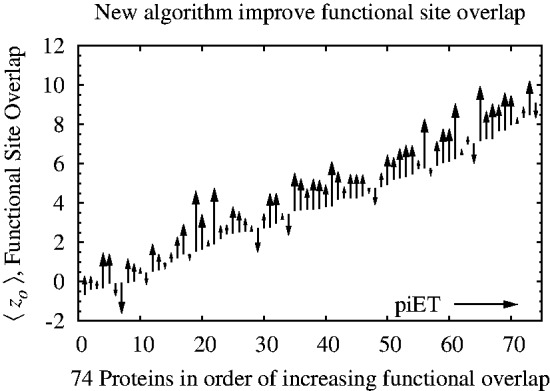
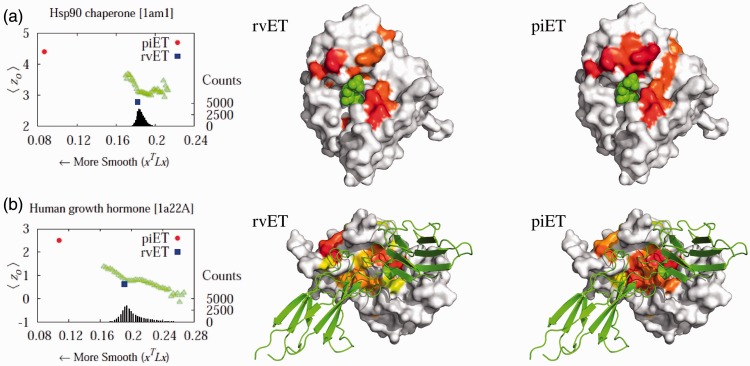
We find that when residue scores of evolutionary importance are distributed smoothly between nearby residues, functional site prediction accuracy improves. Accordingly, we designed a novel measure of evolutionary importance that focuses on the interaction between pairs of structurally neighboring residues. This measure that we term pair-interaction Evolutionary Trace yields greater functional site overlap and better structure-based proteome-wide functional predictions.
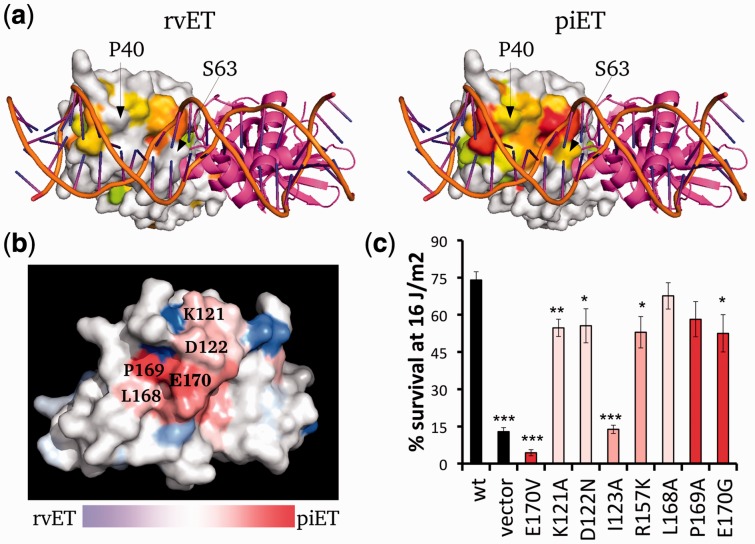
Our data show that the structural smoothness of evolutionary importance is a fundamental feature of the coevolution of sequence, structure and function. Mutations operate on individual residues, but selective pressure depends in part on the extent to which a mutation perturbs interactions with neighboring residues. In practice, this principle led us to redefine the importance of a residue in terms of the importance of its epistatic interactions with neighbors, yielding better annotation of functional residues, motivating experimental validation of a novel functional site in LexA and refining protein function prediction.
Supplementary data are available at Bioinformatics online.
序列、结构和功能共同进化的约束条件尚未完全理解。揭示这种相互关系可以揭示结合、催化和变构的分子基础,从而确定功能并合理指导蛋白质重新设计。这些关系的基础是当蛋白质突变的后果由其发生的遗传背景决定时,会发生上位相互作用。基于先前的数据,我们假设上位力在结构中附近的残基之间作用最强,导致结构中进化重要性的平滑分布。
我们发现,当进化重要性的残基得分在附近残基之间平滑分布时,功能位点预测的准确性会提高。因此,我们设计了一种新的进化重要性度量方法,该方法专注于结构上相邻残基之间的相互作用。我们将这种度量方法称为对相互作用进化痕迹(pair-interaction Evolutionary Trace),它可以提高功能位点的重叠度,并改善基于结构的全蛋白质组功能预测。
我们的数据表明,进化重要性的结构平滑性是序列、结构和功能共同进化的一个基本特征。突变作用于单个残基,但选择压力部分取决于突变对与相邻残基相互作用的干扰程度。实际上,这一原则使我们能够根据残基与其相邻残基的上位相互作用的重要性来重新定义残基的重要性,从而更好地注释功能残基,激励对 LexA 中一个新功能位点的实验验证,并改进蛋白质功能预测。
补充材料可在“生物信息学”在线获取。