Department of Automation, MOE Key Laboratory of Bioinformatics; Bioinformatics Division and Center for Synthetic & Systems Biology, TNLIST, Tsinghua University, Beijing 100084, China.
BMC Med Genomics. 2013 Dec 18;6:57. doi: 10.1186/1755-8794-6-57.
The identification of genes involved in human complex diseases remains a great challenge in computational systems biology. Although methods have been developed to use disease phenotypic similarities with a protein-protein interaction network for the prioritization of candidate genes, other valuable omics data sources have been largely overlooked in these methods.
With this understanding, we proposed a method called BRIDGE to prioritize candidate genes by integrating disease phenotypic similarities with such omics data as protein-protein interactions, gene sequence similarities, gene expression patterns, gene ontology annotations, and gene pathway memberships. BRIDGE utilizes a multiple regression model with lasso penalty to automatically weight different data sources and is capable of discovering genes associated with diseases whose genetic bases are completely unknown.
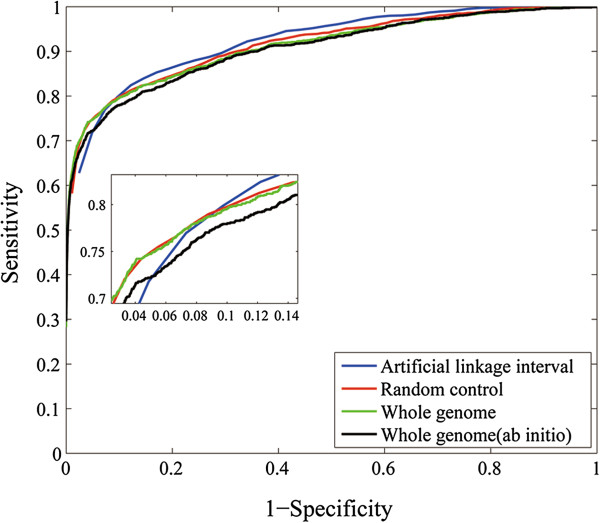
We conducted large-scale cross-validation experiments and demonstrated that more than 60% known disease genes can be ranked top one by BRIDGE in simulated linkage intervals, suggesting the superior performance of this method. We further performed two comprehensive case studies by applying BRIDGE to predict novel genes and transcriptional networks involved in obesity and type II diabetes.
The proposed method provides an effective and scalable way for integrating multi omics data to infer disease genes. Further applications of BRIDGE will be benefit to providing novel disease genes and underlying mechanisms of human diseases.
在计算系统生物学中,鉴定涉及人类复杂疾病的基因仍然是一个巨大的挑战。尽管已经开发了一些方法,利用疾病表型与蛋白质-蛋白质相互作用网络的相似性来优先考虑候选基因,但在这些方法中,其他有价值的组学数据源在很大程度上被忽视了。
基于这一理解,我们提出了一种名为 BRIDGE 的方法,通过整合疾病表型相似性与蛋白质-蛋白质相互作用、基因序列相似性、基因表达模式、基因本体注释和基因途径成员等组学数据,来优先考虑候选基因。BRIDGE 利用带有套索惩罚的多元回归模型自动为不同的数据源赋权,并且能够发现与遗传基础完全未知的疾病相关的基因。
我们进行了大规模的交叉验证实验,结果表明,在模拟的连锁区间内,超过 60%的已知疾病基因可以通过 BRIDGE 排名第一,这表明了该方法的优越性能。我们进一步通过应用 BRIDGE 来预测肥胖和 2 型糖尿病中涉及的新型基因和转录网络,进行了两项全面的案例研究。
该方法为整合多组学数据推断疾病基因提供了一种有效且可扩展的方法。BRIDGE 的进一步应用将有助于提供新的疾病基因和人类疾病的潜在机制。