Rasmussen David A, Volz Erik M, Koelle Katia
Biology Department, Duke University, Durham, North Carolina, United States of America.
Department of Infectious Disease Epidemiology, Imperial College London, London, United Kingdom.
PLoS Comput Biol. 2014 Apr 17;10(4):e1003570. doi: 10.1371/journal.pcbi.1003570. eCollection 2014 Apr.
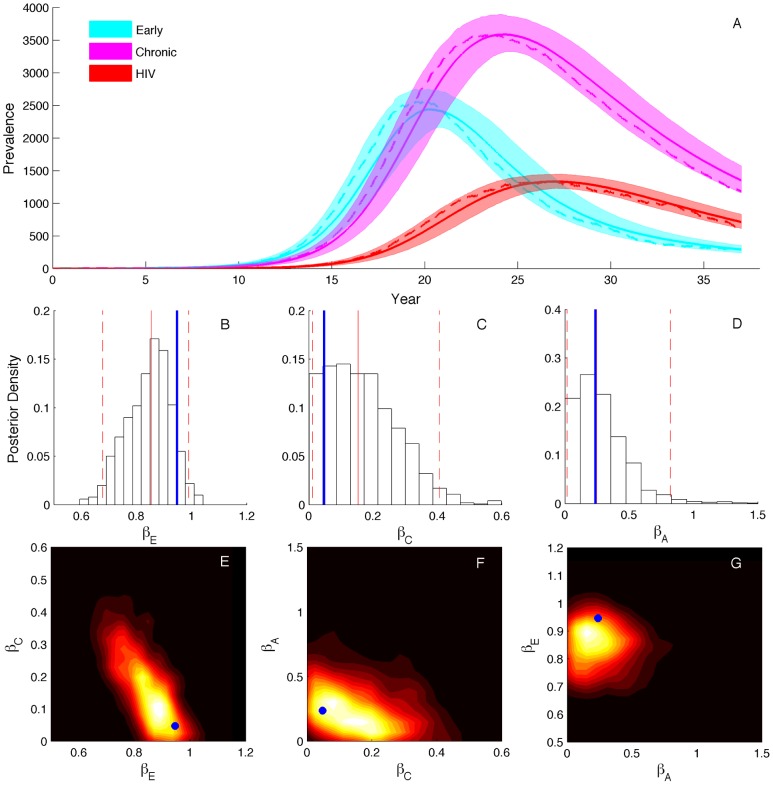
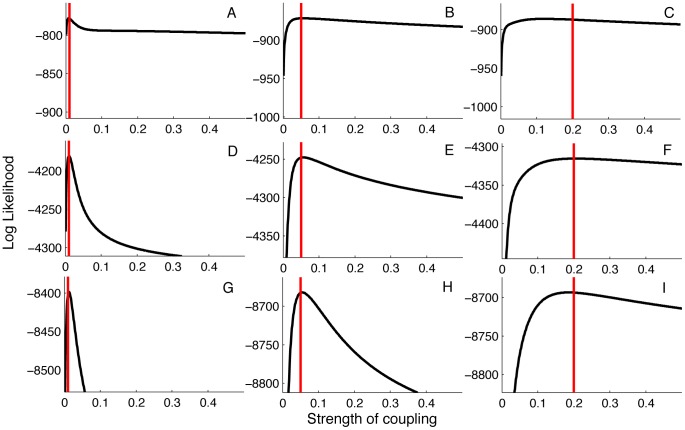
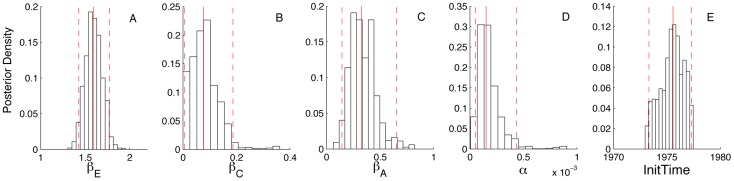
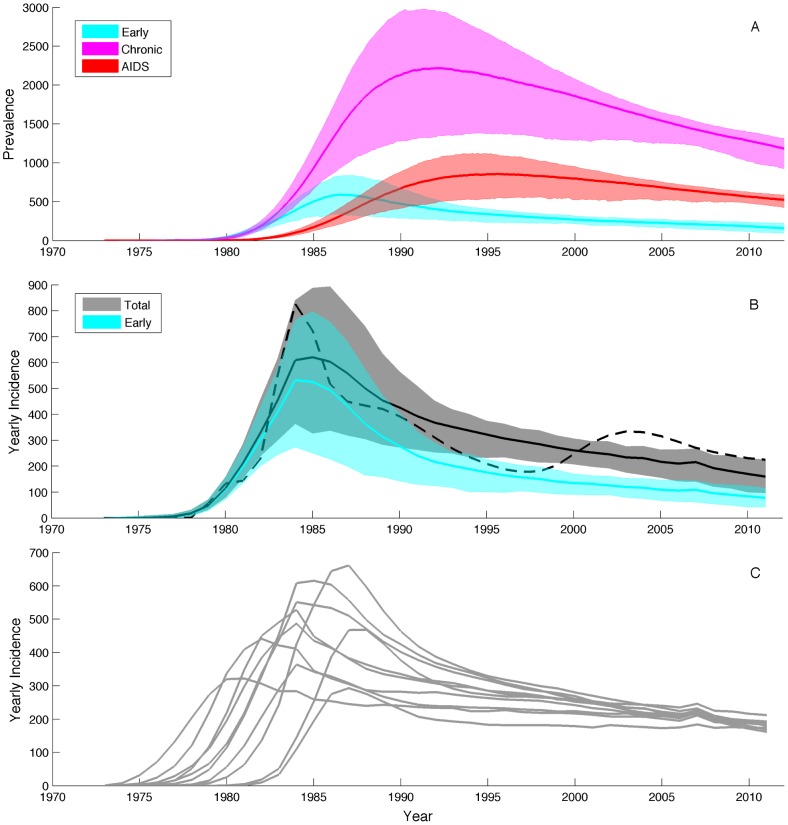
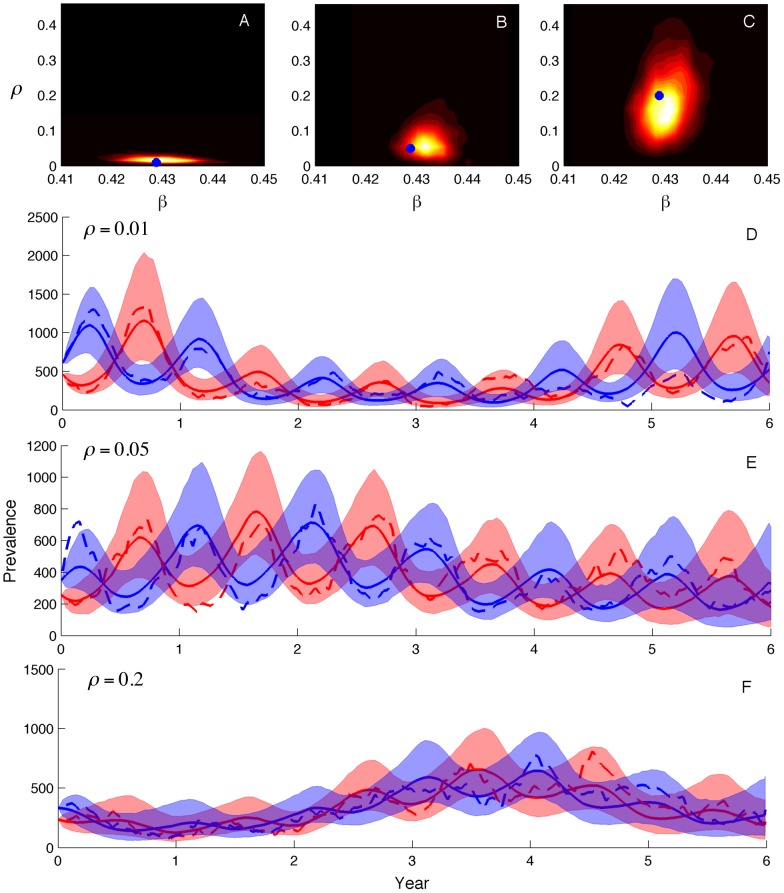
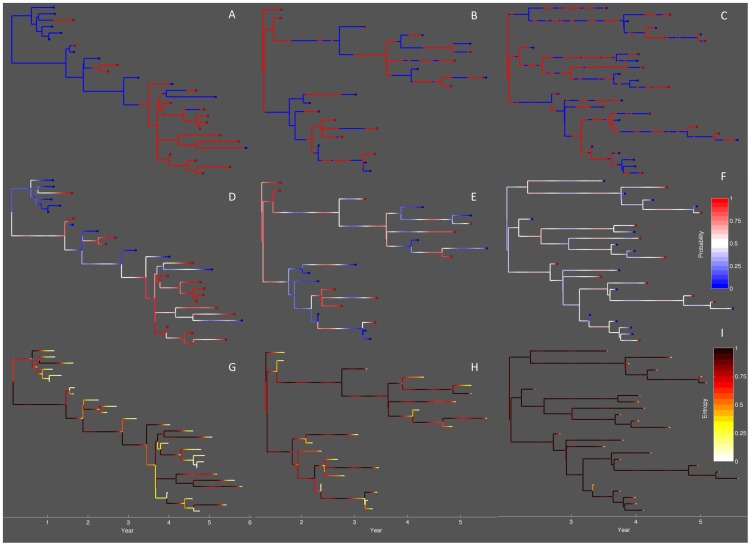
Coalescent theory is routinely used to estimate past population dynamics and demographic parameters from genealogies. While early work in coalescent theory only considered simple demographic models, advances in theory have allowed for increasingly complex demographic scenarios to be considered. The success of this approach has lead to coalescent-based inference methods being applied to populations with rapidly changing population dynamics, including pathogens like RNA viruses. However, fitting epidemiological models to genealogies via coalescent models remains a challenging task, because pathogen populations often exhibit complex, nonlinear dynamics and are structured by multiple factors. Moreover, it often becomes necessary to consider stochastic variation in population dynamics when fitting such complex models to real data. Using recently developed structured coalescent models that accommodate complex population dynamics and population structure, we develop a statistical framework for fitting stochastic epidemiological models to genealogies. By combining particle filtering methods with Bayesian Markov chain Monte Carlo methods, we are able to fit a wide class of stochastic, nonlinear epidemiological models with different forms of population structure to genealogies. We demonstrate our framework using two structured epidemiological models: a model with disease progression between multiple stages of infection and a two-population model reflecting spatial structure. We apply the multi-stage model to HIV genealogies and show that the proposed method can be used to estimate the stage-specific transmission rates and prevalence of HIV. Finally, using the two-population model we explore how much information about population structure is contained in genealogies and what sample sizes are necessary to reliably infer parameters like migration rates.
溯祖理论通常用于从谱系中估计过去的种群动态和人口统计学参数。虽然早期的溯祖理论研究仅考虑简单的人口模型,但理论上的进展使得越来越复杂的人口情景得以考虑。这种方法的成功促使基于溯祖的推断方法应用于种群动态快速变化的群体,包括RNA病毒等病原体。然而,通过溯祖模型将流行病学模型拟合到谱系仍然是一项具有挑战性的任务,因为病原体群体通常表现出复杂的非线性动态,并且由多种因素构成结构。此外,在将此类复杂模型拟合到实际数据时,通常有必要考虑种群动态中的随机变化。利用最近开发的能够适应复杂种群动态和种群结构的结构化溯祖模型,我们开发了一个统计框架,用于将随机流行病学模型拟合到谱系。通过将粒子滤波方法与贝叶斯马尔可夫链蒙特卡罗方法相结合,我们能够将广泛的具有不同种群结构形式的随机、非线性流行病学模型拟合到谱系。我们使用两个结构化流行病学模型展示了我们的框架:一个具有感染多阶段疾病进展的模型和一个反映空间结构的双种群模型。我们将多阶段模型应用于HIV谱系,并表明所提出的方法可用于估计特定阶段的传播率和HIV的流行率。最后,使用双种群模型,我们探讨了谱系中包含了多少关于种群结构的信息,以及可靠推断迁移率等参数所需的样本量。