Azam Faizul, Amer Abdualrahman M, Abulifa Abdullah R, Elzwawi Mustafa M
Faculty of Pharmacy, Misurata University, Misurata, Libya ; Department of Pharmaceutical Chemistry, Nims Institute of Pharmacy, Nims University, Jaipur, Rajasthan, India.
Faculty of Pharmacy, Misurata University, Misurata, Libya.
Drug Des Devel Ther. 2014 Oct 23;8:2045-59. doi: 10.2147/DDDT.S67778. eCollection 2014.
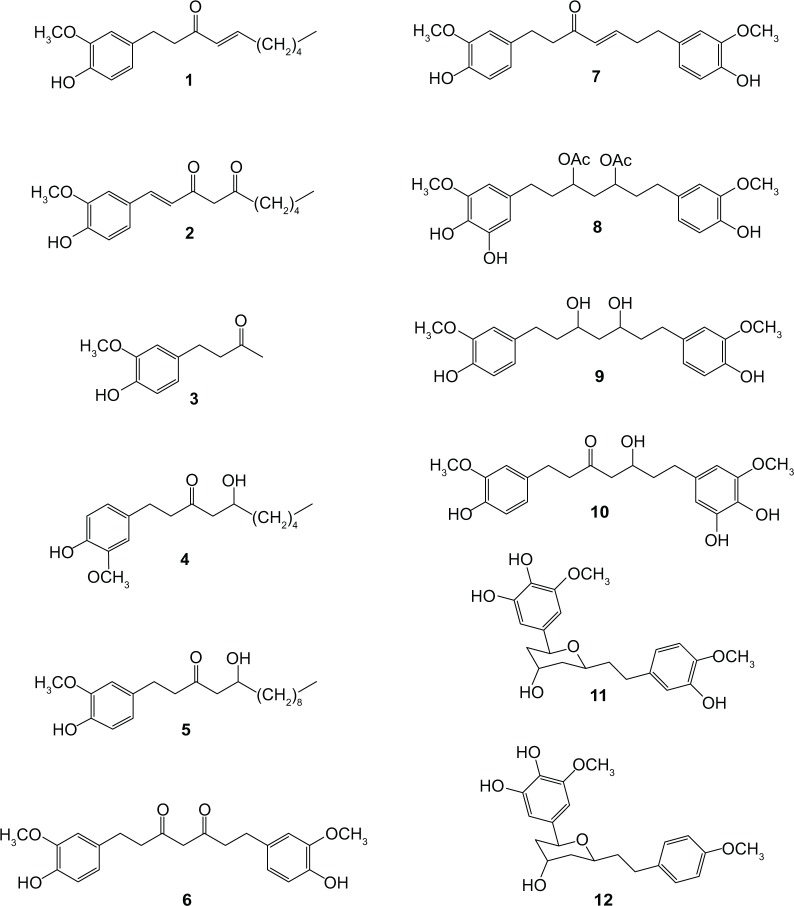
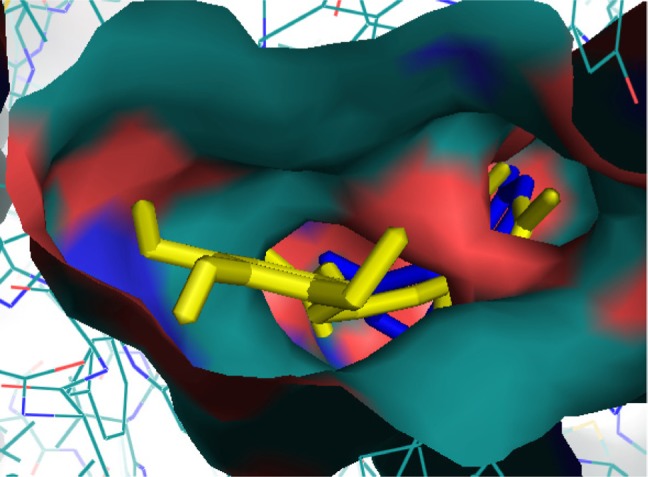
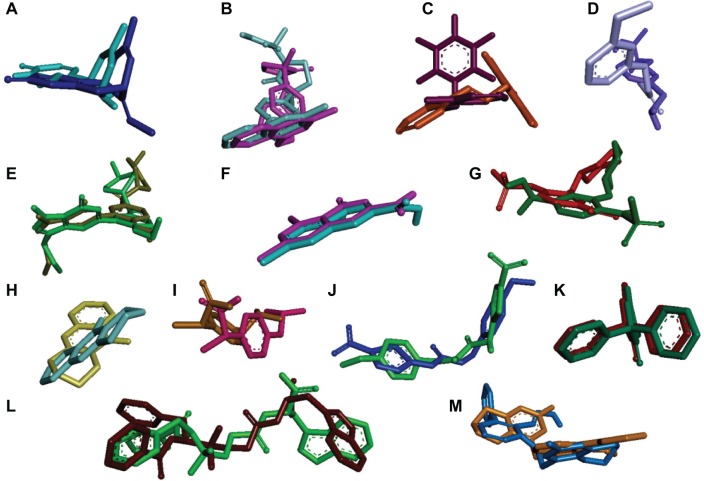
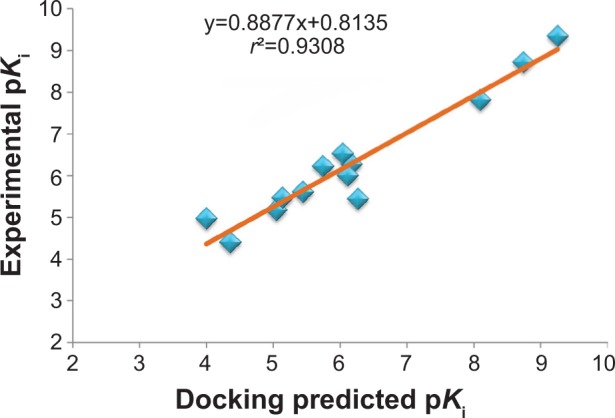
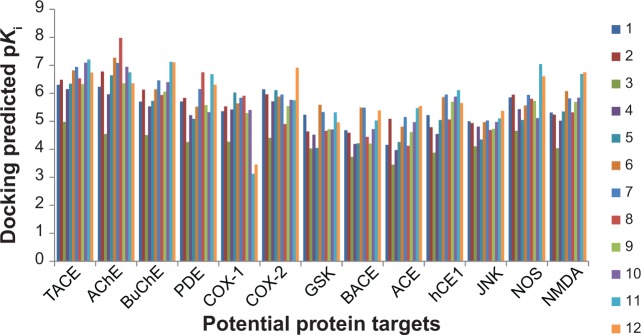
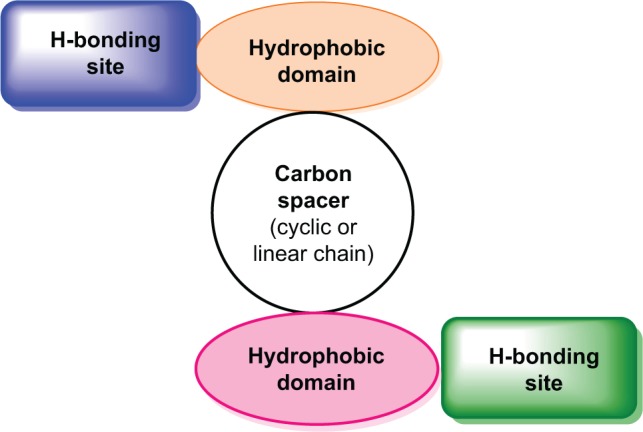
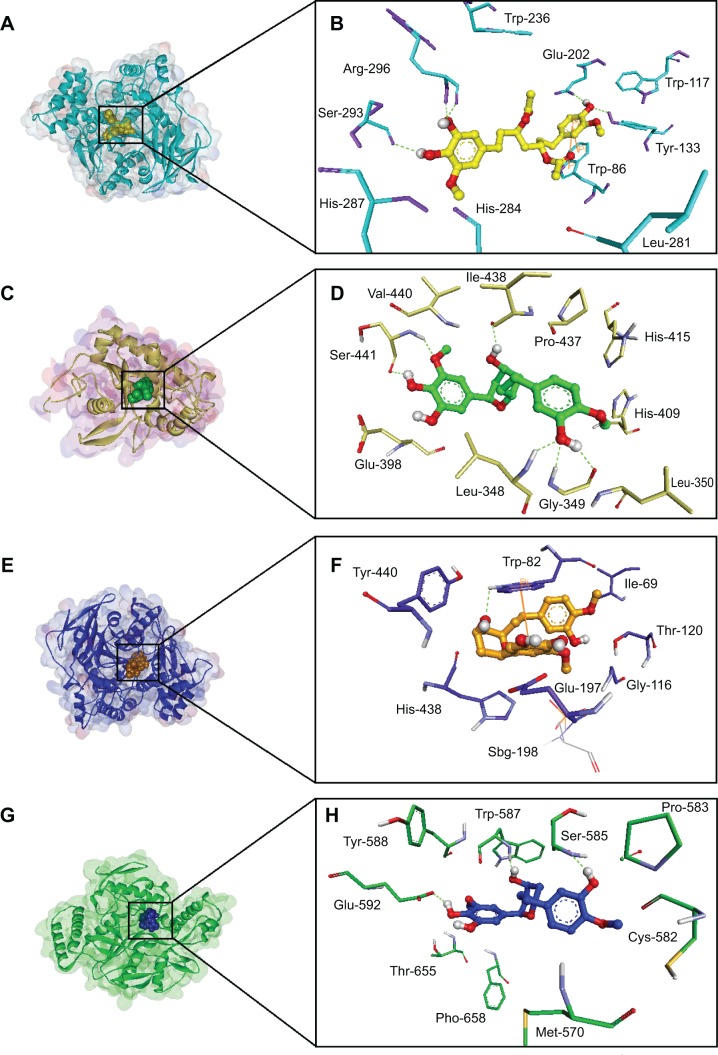
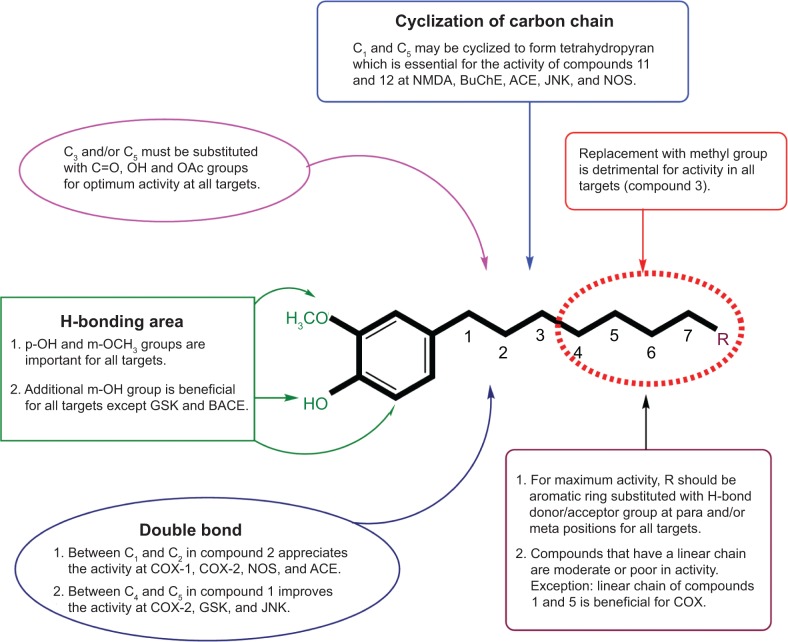
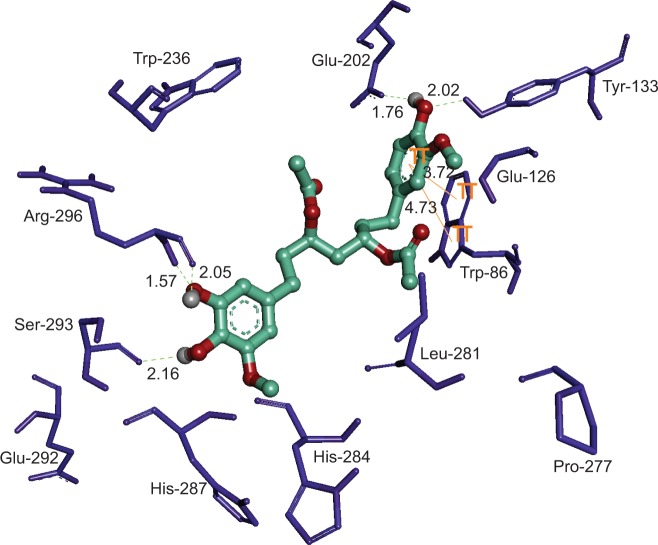
Ginger (Zingiber officinale), despite being a common dietary adjunct that contributes to the taste and flavor of foods, is well known to contain a number of potentially bioactive phytochemicals having valuable medicinal properties. Although recent studies have emphasized their benefits in Alzheimer's disease, limited information is available on the possible mechanism by which it renders anti-Alzheimer activity. Therefore, the present study seeks to employ molecular docking studies to investigate the binding interactions between active ginger components and various anti-Alzheimer drug targets. Lamarckian genetic algorithm methodology was employed for docking of 12 ligands with 13 different target proteins using AutoDock 4.2 program. Docking protocol was validated by re-docking of all native co-crystallized ligands into their original binding cavities exhibiting a strong correlation coefficient value (r (2)=0.931) between experimentally reported and docking predicted activities. This value suggests that the approach could be a promising computational tool to aid optimization of lead compounds obtained from ginger. Analysis of binding energy, predicted inhibition constant, and hydrophobic/hydrophilic interactions of ligands with target receptors revealed acetylcholinesterase as most promising, while c-Jun N-terminal kinase was recognized as the least favorable anti-Alzheimer's drug target. Common structural requirements include hydrogen bond donor/acceptor area, hydrophobic domain, carbon spacer, and distal hydrophobic domain flanked by hydrogen bond donor/acceptor moieties. In addition, drug-likeness score and molecular properties responsible for a good pharmacokinetic profile were calculated by Osiris property explorer and Molinspiration online toolkit, respectively. None of the compounds violated Lipinski's rule of five, making them potentially promising drug candidates for the treatment of Alzheimer's disease.
生姜(姜科植物姜)虽是一种常见的饮食佐料,能为食物增添味道和风味,但众所周知其含有多种具有潜在生物活性的植物化学物质,具备宝贵的药用特性。尽管近期研究强调了它们在阿尔茨海默病中的益处,但关于其发挥抗阿尔茨海默病活性的可能机制的信息却很有限。因此,本研究旨在通过分子对接研究来探究生姜活性成分与各种抗阿尔茨海默病药物靶点之间的结合相互作用。使用拉马克遗传算法方法,借助AutoDock 4.2程序将12种配体与13种不同的靶蛋白进行对接。通过将所有天然共结晶配体重新对接至其原始结合腔来验证对接方案,实验报道的活性与对接预测的活性之间呈现出很强的相关系数值(r (2)=0.931)。该值表明该方法可能是一种很有前景的计算工具,有助于优化从生姜中获得的先导化合物。对配体与靶受体的结合能、预测抑制常数以及疏水/亲水相互作用的分析表明,乙酰胆碱酯酶是最有前景的,而c-Jun氨基末端激酶被认为是最不理想的抗阿尔茨海默病药物靶点。共同的结构要求包括氢键供体/受体区域、疏水区、碳间隔基以及两侧带有氢键供体/受体基团的远端疏水区。此外,分别通过Osiris性质探索器和Molinspiration在线工具包计算了类药性质得分以及负责良好药代动力学特征的分子性质。这些化合物均未违反Lipinski的五规则,使其有可能成为治疗阿尔茨海默病的有前景的候选药物。