Fiannaca Antonino, La Rosa Massimo, La Paglia Laura, Rizzo Riccardo, Urso Alfonso
BMC Bioinformatics. 2015;16 Suppl 4(Suppl 4):S7. doi: 10.1186/1471-2105-16-S4-S7. Epub 2015 Feb 23.
MicroRNAs (miRNAs) are important key regulators in multiple cellular functions, due to their a crucial role in different physiological processes. MiRNAs are differentially expressed in specific tissues, during specific cell status, or in different diseases as tumours. RNA sequencing (RNA-seq) is a Next Generation Sequencing (NGS) method for the analysis of differential gene expression. Using machine learning algorithms, it is possible to improve the functional significance interpretation of miRNA in the analysis and interpretation of data from RNA-seq. Furthermore, we tried to identify some patterns of deregulated miRNA in human breast cancer (BC), in order to give a contribution in the understanding of this type of cancer at the molecular level.
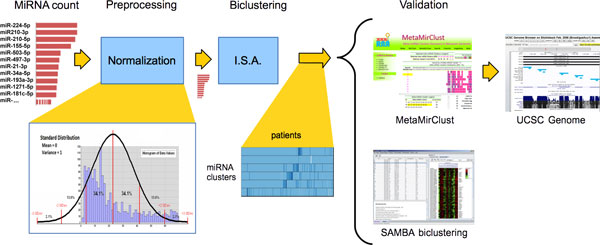
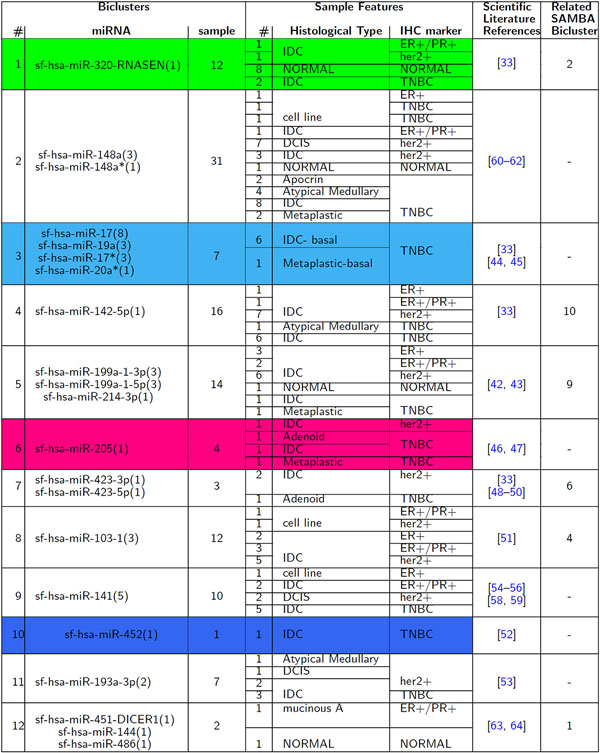
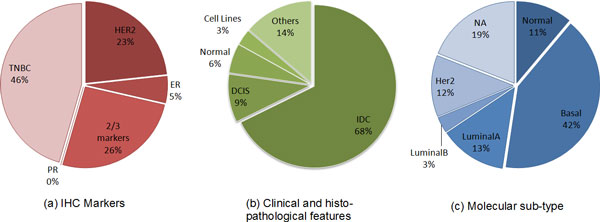
We adopted a biclustering approach, using the Iterative Signature Algorithm (ISA) algorithm, in order to evaluate miRNA deregulation in the context of miRNA abundance and tissue heterogeneity. These are important elements to identify miRNAs that would be useful as prognostic and diagnostic markers. Considering a real word breast cancer dataset, the evaluation of miRNA differential expressions in tumours versus healthy tissues evidenced 12 different miRNA clusters, associated to specific groups of patients. The identified miRNAs were deregulated in breast tumours compared to healthy controls. Our approach has shown the association between specific sub-class of tumour samples having the same immuno-histo-chemical and/or histological features. Biclusters have been validated by means of two online repositories, MetaMirClust database and UCSC Genome Browser, and using another biclustering algorithm.
The obtained results with biclustering algorithm aimed first of all to give a contribute in the differential expression analysis in a cohort of BC patients and secondly to support the potential role that these non-coding RNA molecules could play in the clinical practice, in terms of prognosis, evolution of tumour and treatment response.
微小RNA(miRNA)是多种细胞功能的重要关键调节因子,因为它们在不同生理过程中起着至关重要的作用。miRNA在特定组织、特定细胞状态或不同疾病(如肿瘤)中差异表达。RNA测序(RNA-seq)是一种用于分析差异基因表达的新一代测序(NGS)方法。使用机器学习算法,可以在RNA-seq数据分析和解释中提高对miRNA功能意义的解读。此外,我们试图识别人类乳腺癌(BC)中失调miRNA的一些模式,以便在分子水平上为理解这类癌症做出贡献。
我们采用了一种双聚类方法,使用迭代特征算法(ISA)算法,以在miRNA丰度和组织异质性的背景下评估miRNA失调情况。这些是识别可用作预后和诊断标志物的miRNA的重要因素。考虑一个真实的乳腺癌数据集,肿瘤组织与健康组织中miRNA差异表达的评估显示了12个不同的miRNA簇,与特定患者群体相关。与健康对照相比,所鉴定的miRNA在乳腺肿瘤中失调。我们的方法显示了具有相同免疫组织化学和/或组织学特征的特定肿瘤样本亚类之间的关联。双聚类已通过两个在线数据库(MetaMirClust数据库和UCSC基因组浏览器)以及使用另一种双聚类算法进行了验证。
双聚类算法获得的结果首先旨在为一组BC患者的差异表达分析做出贡献,其次是支持这些非编码RNA分子在临床实践中在预后、肿瘤进展和治疗反应方面可能发挥的潜在作用。