Chen Hui, Liu Ying, Zhang Menghui, Wang Guoyang, Qi Zhengnan, Bridgewater Laura, Zhao Liping, Tang Zisheng, Pang Xiaoyan
State Key Laboratory of Microbial Metabolism, School of Life Sciences and Biotechnology, Shanghai Jiao Tong University, Shanghai 200240, China.
Department of Endodontics, Ninth People's Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai Key Laboratory of Stomatology, Shanghai 200011, China.
Sci Rep. 2015 Mar 12;5:9053. doi: 10.1038/srep09053.
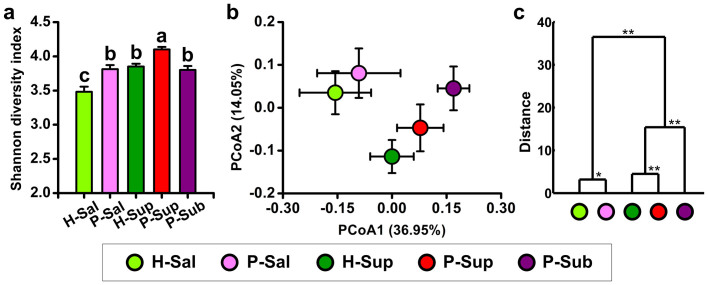
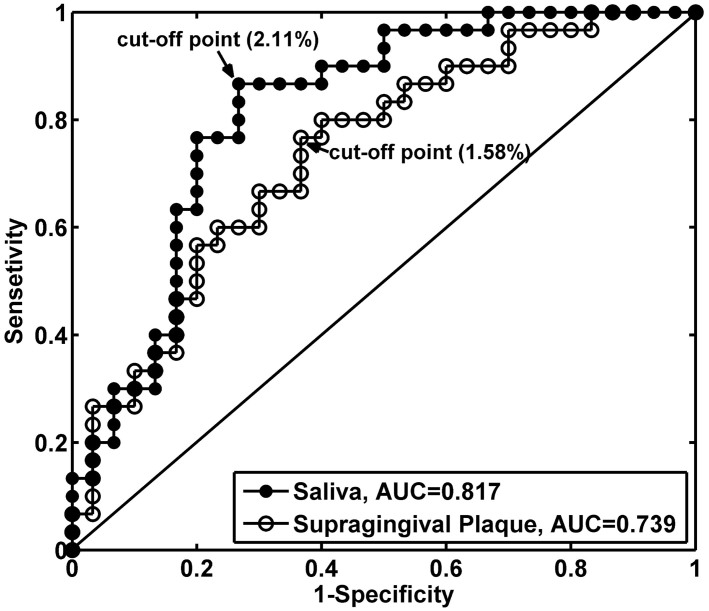
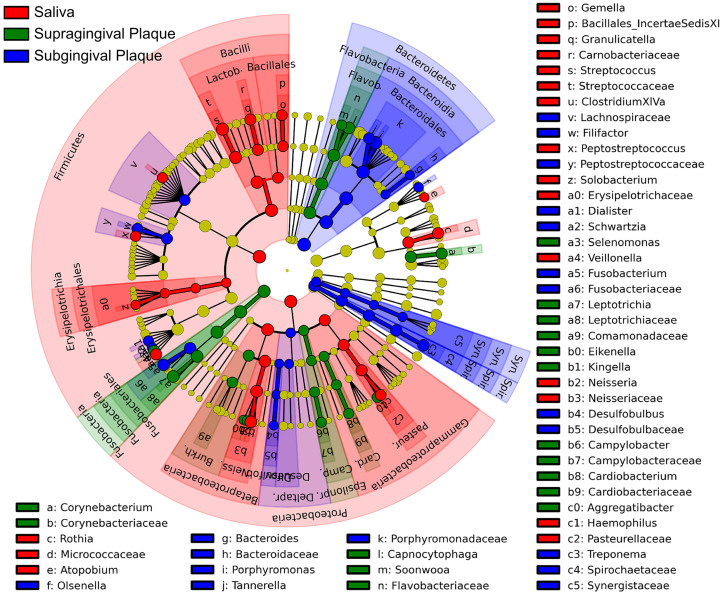
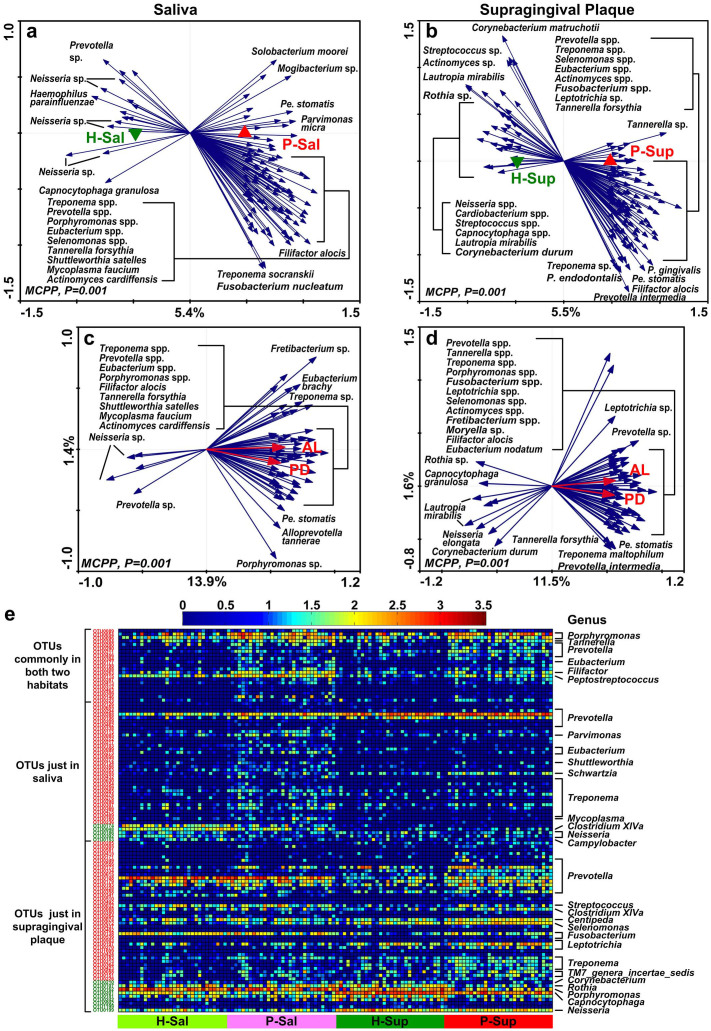
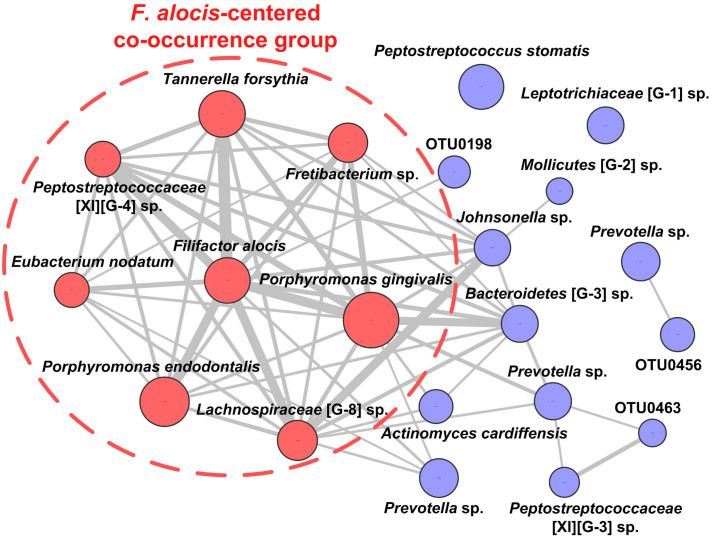
Periodontitis is a highly prevalent polymicrobial disease worldwide, yet the synergistic pattern of the multiple oral pathogens involved is still poorly characterized. Here, saliva, supragingival and subgingival plaque samples from periodontitis patients and periodontally healthy volunteers were collected and profiled with 16S rRNA gene pyrosequencing. Different oral habitats harbored significantly different microbiota, and segregation of microbiota composition between periodontitis and health was observed as well. Two-step redundancy analysis identified twenty-one OTUs, including Porphyromonas gingivalis, Tannerella forsythia and Filifactor alocis, as potential pathogens that were significantly associated with periodontitis and with two periodontitis diagnostic parameters (pocket depth and attachment loss) in both saliva and supragingival plaque habitats. Interestingly, pairwise correlation analysis among the 21 OTUs revealed that Filifactor alocis was positively correlated with seven other putative pathogens (R > 0.6, P < 0.05), forming a co-occurrence group that was remarkably enriched in all three habitats of periodontitis patients. This bacterial cluster showed a higher diagnostic value for periodontitis than did any individual potential pathogens, especially in saliva. Thus, our study identified a potential synergistic ecological pattern involving eight co-infecting pathogens across various oral habitats, providing a new framework for understanding the etiology of periodontitis and developing new diagnoses and therapies.
牙周炎是一种在全球范围内高度流行的多微生物疾病,但所涉及的多种口腔病原体的协同模式仍未得到充分描述。在这里,收集了牙周炎患者和牙周健康志愿者的唾液、龈上和龈下菌斑样本,并用16S rRNA基因焦磷酸测序进行分析。不同的口腔生境中存在着显著不同的微生物群,同时也观察到了牙周炎患者与健康者之间微生物群组成的差异。两步冗余分析确定了21个操作分类单元(OTU),包括牙龈卟啉单胞菌、福赛坦氏菌和具核梭杆菌,它们是与牙周炎以及唾液和龈上菌斑生境中的两个牙周炎诊断参数(牙周袋深度和附着丧失)显著相关的潜在病原体。有趣的是,对这21个OTU进行的成对相关性分析显示,具核梭杆菌与其他七种假定病原体呈正相关(R > 0.6,P < 0.05),形成了一个在牙周炎患者的所有三个生境中均显著富集的共现组。该细菌簇对牙周炎的诊断价值高于任何单个潜在病原体,尤其是在唾液中。因此,我们的研究确定了一种潜在的协同生态模式,涉及跨越各种口腔生境的八种共同感染病原体,为理解牙周炎的病因以及开发新的诊断方法和治疗手段提供了一个新的框架。